Congenital malformations may affect any part of the eye and the ocular adnexa. Developmental defects may occur in isolation or as part of a larger systemic malformation syndrome. Many malformations can severely impair vision, whereas others have only cosmetic significance, and still others cause no symptoms and may go undiscovered or may be noted incidentally on routine eye examination. Congenital anomalies have numerous causes, most commonly of developmental genetic origin. The genetic basis of congenital eye and orbit anomalies is just beginning to be delineated, and future research on the subject will undoubtedly broaden understanding of the developmental etiology, pathophysiology, and treatment of congenital ocular disorders.
Malformations of the eye, orbit, and ocular adnexa are known to occur in isolation, in combination, or as part of a larger systemic malformation syndrome. Many malformations can severely impair vision, others have only cosmetic significance, and still others cause no symptoms and may go undiscovered or may be noted incidentally on routine eye examination . Congenital anomalies have various causes. The most common causes are defects in genes critical to normal development, but also include aneuploidies, in-utero exposure to exogenous teratogens (ie, drugs or infectious microorganisms), and obstetrical complication (ie, oligohydramnios) . Both germline and somatic mutations can cause congenital malformations. Much of the advancement in the field of dysmorphology and in the understanding of congenital malformations in recent years is the result of progress with the genome projects (including the human genome project), which have broadened understanding of the genes implicated in eye and orbit development, and provided a genetic basis for many human developmental eye diseases, including many well-known syndromes with malformed eyes and orbits . Four categories of congenital anomalies have been proposed, including those resulting from (1) single morphogenetic defects, (2) intrauterine mechanical constraint on an otherwise normal embryo or fetus, (3) destruction of a normal structure, and (4) dysplasia, defined as a defect in the differentiation and organization of a tissue. Many of these dysplasias are caused by a single morphogenetic anomaly in development that leads to a subsequent series of defects, defining a sequence or syndrome .
Embryology of the human eye
The major structures of the eye are derived from ectoderm, both neural and surface. The sequence of steps that follow have been presented in much greater detail with illustrations elsewhere . The optic pits are formed at 22 days’ gestation and then extend outward to form the optic vesicles at 26 to 27 days’ gestation. These then contact the overlying surface ectoderm, causing both layers to invaginate. The optic vesicles then form the optic cups at about 4 weeks’ gestation, with the inner layer forming the neural (or neurosensory) retina and the outer layer forming the pigmented retina (or pigmented retinal epithelium). The lens placode and lens vesicle then form through a process of invagination and pinching off of surface ectoderm. This process also contributes to formation of the presumptive cornea .
The retina begins to differentiate very early in embryogenesis. At 26 days, three to four rows of cells are present, and by 32 to 33 days, five to six rows line the inner layer of the optic cup. Maturation and melanization of the retinal pigment epithelium begins posteriorly at the site of the presumptive macula and proceeds peripherally . The ganglion cells are produced first, followed by cone photoreceptors, horizontal and amacrine cells, later rod photoreceptors, and finally bipolar cells and Muller cells .
Primary vitreous, a highly vascularized gel, develops between weeks 4 and 6, and is then replaced with the avascular secondary vitreous and finally the tertiary vitreous (the suspensory zonules) by the fourth month of gestation . The optic cup, which is connected to the developing forebrain by the optic stalk, contains an inferior and ventral fold, denoted the embryonic fissure , through which mesenchymal and vascular tissues, including the hyaloid artery and a branch of the ophthalmic artery, enter the globe. Closure of this fissure between the fifth and seventh week through cell division and apoptosis is critical to normal iris development. Incomplete closure leads to colobomas of the iris, retina, or choroid . The vascular system of the eye and orbit is derived from the primitive internal carotid system, and the primitive dorsal ophthalmic artery supplies the entire eye and orbit at this time .
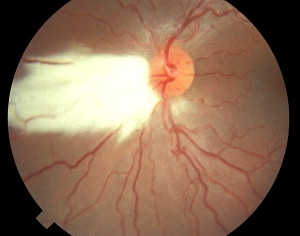
Melanogenesis at optic fissure closure results in vacuolation of the inner layers of the optic stalk to accommodate nerve fibers sent out by differentiating retinal neurons. Immigration of retinal ganglion cell axons results in optic nerve formation . Pruning of the initial overproduction of axons results in confirmed connections and the elimination of nonfunctional processes; approximately 1.2 million active fibers remain with some crossover at the optic chiasm. Myelination of the optic nerve occurs between 5 and 8 months’ gestation and proceeds from the geniculate bodies to the globe. Ectopic myelination of the nerve fiber layer within the eye can be seen in some patients as a flat, feathery, glossy white sheen on the surface of the retina ( Fig. 1 ) .
Cornea and iris form at about 6 weeks’ gestation. The first of three waves of mesenchymal cell migrations produces the corneal inner epithelium and the trabecular meshwork, the second results in formation of the corneal stroma, and the third forms the iris stroma. Two anterior chamber structures, the angle and the ciliary body, also develop at approximately 6 weeks’ gestation. Lens development is hastened at this point by atrophy of the hyaloid artery and closure of the lumen of the lens vesicle by 7 weeks’ gestation . The lens, derived from the lens epithelium and developed by formation of primary and secondary lens fibers, begins to reduce DNA synthesis at its posterior pole, resulting in cell elongation and the development of an organized series of crystallins and other lens proteins . Choroid and sclera are formed from condensations of mesenchymal neural crest cells .
Extraocular muscles develop from masses of paraxial mesoderm during weeks 4 to 5, with scleral insertion by the second month of gestation. Levator palpebrae superioris is the last striated extraocular muscle to form, occurring during the third month of gestation. The eyelids develop from ectoderm, mesoderm, and, in the case of the tarsus and connective tissue of the eyelid, mesenchymal neural crest cells . These structures begin to differentiate at the sixth week of gestation, fusing at the eighth week. The eyelids begin to separate around the fifth month, beginning nasally and extending temporally, with complete separation by the seventh month . The lacrimal glands are formed from epithelial buds arising from the basal epithelial layer of the conjunctiva. Canalization of the lacrimal duct begins at the third month of gestation and is usually completed by the sixth month, but a persistent closure of the lower portion of the duct at birth may result in congenital nasolacrimal duct obstruction. The dilator and sphincter muscles of the iris develop from neuroectoderm between the sixth and eighth months of gestation, and iris vasculature stems from mesoderm .
The orbit, which is formed by seven bones, including the maxillary, zygomatic, frontal, sphenoid, palatine, ethmoid, and lacrimal, undergoes rapid changes in size and shape beginning at 6 months’ gestation. The orbital diameter begins to increase rapidly at this time, after transitioning from a previous linear growth curve .
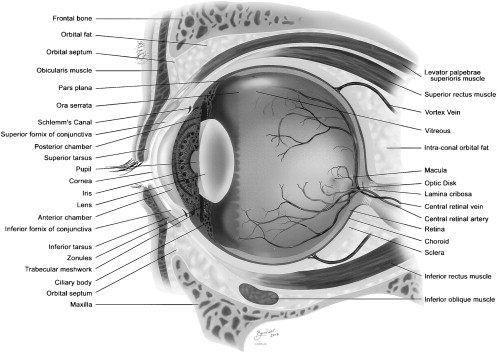
Genetic regulation is critical to successful ocular embryogenesis. The two genes that have been described as most important in ocular development are the PAX6 gene (chromosome 11p13) and the Rx gene (chromosome 18). Both genes belong to a large family of factors that are related to the homeodomain region of the Drosophila paired protein. Early induction of both genes causes a series of gene activations and depressions which are tantamount to normal development of the mature eye. Rx and PAX6 are both expressed in proliferating cells. PAX6 is also expressed in surface ectoderm of the lens primordia. Mutations of PAX6 have been shown to lead to aniridia, congenital cataract, Peter’s anomaly, and midline fusion defects. Absence of the gene leads to anophthalmia. Rx is associated with retinal proliferation . Other genes that are believed to be involved in ocular development include FOXC1 and PITX2 genes, mutations of which have been described in Axenfeld-Rieger syndrome . Normal eye anatomy is depicted in Fig. 2 .
Congenital abnormalities and syndromes of the eye
Abnormalities of the anterior segment of the eye
Anterior segment dysgenesis
A significant number of congenital ocular anomalies, all of them rare, involve the anterior segment of the eye, which includes the cornea, iris, ciliary body, and lens. The most common of these uncommon disorders is termed the Axenfeld-Rieger spectrum of anomalies , which includes several abnormalities, such as posterior embryotoxon (an arcuate white line in the periphery of the cornea), abnormalities of pupillary shape or location, polycoria (full-thickness defects of the iris independent of pupillary defects), and adhesions between the iris and cornea. These ocular abnormalities may coexist with a spectrum of extraocular anomalies as part of an autosomal dominant multisystem congenital disorder that includes facial dysmorphism, redundant periumbilical skin, and anomalies of dentition. Many patients who have Axenfeld-Rieger develop glaucoma, and therefore should be periodically screened for this condition .
Peter’s anomaly
Children born with a characteristic white opacity of the cornea are classified as having Peter’s anomaly. The opacity is usually, but not necessarily, an avascular, central defect that causes a significant visual disturbance. Patients often also exhibit iridocorneal adhesions with or without an anteriorly displaced cataractous lens. Peter’s anomaly may be unilateral or bilateral, and patients are predisposed to glaucoma. In the absence of absolute contraindications, surgery for Peter’s anomaly, which includes corneal transplantation with or without lens extraction, must proceed at an early age to avoid irreversible amblyopia resulting from visual deprivation in the first weeks of life. An alternative procedure involves the resection of a large section of iris (sector iridectomy) to allow the child to see around the corneal opacity .
Peter’s anomaly is known to occur in isolation, as part of a larger ocular syndrome such as aniridia or microphthalmia, or as part of a systemic syndromic presentation in Peter’s Plus syndrome, involving developmental delay, short stature, and skeletal dysplasia .
Peter’s anomaly is believed to represent a nonheritable disorder and has been associated with mutations in the PAX6 gene and the CYP1B1 gene for congenital glaucoma .
Other congenital opacities of the cornea
Several other corneal opacities are congenital in origin. A primary bilateral, gray-white corneal opacification is seen in congenital hereditary endothelial dystrophy (CHED) involving the innermost corneal layer and in congenital hereditary stromal dystrophy (CHSD) involving the middle layer of the cornea. A primary white vascularized opacification of the cornea is characteristic of sclerocornea; focal white opaque lesions suggest corneal dermoid . Although not typically threatening to the visual axis, dermoids of the corneal limbus, which are small white masses that often have hairs growing from them, may cause amblyopia secondary to astigmatism, may cause ocular discomfort, and are seen in Goldenhar syndrome (oculo–auricular–vertebral spectrum). Goldenhar syndrome results from aberrant development of the first or second branchial arch and includes, in addition to epibulbar dermoids, hemifacial microsomia with resultant orbital asymmetry, small ears with skin tags, and vertebral anomalies .
Corneal clouding may also occur secondary to a host of metabolic storage diseases (although the cornea is often clear at birth), cystinosis, congenital infection, and birth or amniocentesis trauma. Forceps injury to the cornea with rupture of Descemet’s membrane may result in corneal clouding caused by edema. A large-appearing, cloudy cornea is a frequent presentation of congenital glaucoma ( Fig. 3 ) .
Aniridia
Aniridia is a rare ocular disorder characterized by deficiency of iris tissue, ranging from mild hypoplasia to almost total absence of iris. Associated abnormalities include macular and optic nerve hypoplasia (ONH), cataracts, glaucoma and progressive corneal opacification, attendant vision impairment, and nystagmus ( Fig. 4 ).
Aniridia may occur as a familial autosomal dominant disorder or as a sporadic disorder. It may occur in isolation or in association with several systemic abnormalities. In particular, sporadic aniridia is associated with Wilm’s tumor, and often also with genitourinary abnormalities and mental retardation (WAGR), attributed to mutation of the PAX6 gene on chromosome 11p13 .
Persistent pupillary membranes
Incomplete resorption of the normal intrauterine pupillary membrane rarely results in visual disturbance and is more frequently an incidental finding, with small strands projecting freely from the iris into the anterior chamber, or longer strands, even fine webs, spanning the pupil ( Fig. 5 ). Typically, lysis of the strands results from physiologic pupillary constriction and dilation, but pharmacologic dilation may be needed in refractory cases. Surgery is usually indicated only if substantial strands or bands attach to the lens, causing miosis or cataract. Prompt referral to an ophthalmologist is indicated if the persistent pupillary strands obstruct the red reflex . Rarely, patent vessels within pupillary membrane remnants can rupture, causing spontaneous hyphema.
Congenital cysts of the margin of the pupil
Originating in the posterior pigmented epithelium of the iris at the pupillary margin, these bead-like cysts cause the edge of the pupil to appear irregular and impart a brownish appearance which is easily seen when checking for the presence of the red reflex. Because these cysts uncommonly involve the visual axis, referral to an ophthalmologist is rarely indicated. Congenital cysts of the pupil margin typically disappear over time, making surgical removal unnecessary. Pharmacologic dilation of the pupil may be used in refractory cases or cases involving the visual axis .
Physiologic anisocoria
About a quarter of the population has a size discrepancy of up to 2 mm between the two pupils. Physiologic anisocoria requires that the pupils be round, centrally located, and briskly reactive. If the anisocoria is physiologic, as in most cases, the proportional discrepancy in size will remain relatively constant in all light conditions .
Congenital cataract
Congenital cataract occurs in 1:4000 to 1:10,000 newborns ( Fig. 6 ). It may be unilateral or bilateral, an isolated finding, or part of an extensive spectrum of congenital disorders, both heritable and nonheritable, involving chromosomal aberrations; multisystem genetic syndromes; congenital metabolic derangements such as galactosemia; congenital infections; or congenital ocular malformation syndromes such as Peter’s anomaly or aniridia. It is a rare result of birth trauma or amniocentesis injury .
By definition, any lens opacity is considered a cataract. Cataracts that do not involve the visual axis may be visually insignificant. Even small central cataracts (<3 mm) may become visually insignificant with the instillation of an accommodation-sparing dilating drop with or without patching of the unaffected eye. Cataracts are described with respect to the part of the lens that is involved. Anterior opacities (anterior polar) include dot anterior polar cataract, anterior lenticonus often associated with Alport syndrome (caused by a defect of collagen and characterized by renal and, less commonly, cochlear anomalies), anterior pyramidal cataract, anterior subcapsular cataract, and anterior capsular opacity associated with persistent pupillary membrane strands. Posterior opacities include posterior polar cataract, posterior lenticonus, and posterior subcapsular cataract (commonly seen in iritis). Cataracts within the lens include nuclear cataract and lamellar cataract, the latter of which involves individual layers of the interior of the lens. Many genes are responsible for the varying presentations of congenital cataract; autosomal dominant, autosomal recessive, and X-linked recessive transmissions occur .
Treatment for visually significant congenital cataracts should be initiated immediately after detection (usually by the pediatrician’s careful examination of the red reflex, which will appear partially or completely obscured, depending on the size and location of the cataract). Prompt referral to an ophthalmologist is critical for any abnormality of the red reflex, including asymmetry between the eyes and the presence of a white or dark reflex, to establish the diagnosis of congenital cataract or other ocular abnormality and to guide treatment. Urgent referral must be made if congenital cataract is suspected in the first year of life, particularly in the first 6 weeks to 3 months. Although response to treatment diminishes with age, children aged up to 9 to 11 years who have congenital cataract may benefit from treatment .
Treatment of the visually significant cataract that is not amenable to pupillary dilation and patching involves removal of the cataractous lens as soon as possible to avoid irreversible failure of visual development resulting from lack of visual input to the affected eye or eyes. The resultant surgical aphakia is then corrected with glasses, contact lenses, or an intraocular lens implant (IOL) at surgery. Intraocular lens insertion for children aged 2 years and older is becoming increasingly accepted in the absence of contraindications such as iritis . IOL implantation in infants can yield poor results . Patching of the phakic eye is a critical treatment component in unilateral congenital cataract .
Persistent hyperplastic primary vitreous
Lens development in the embryo is hastened between weeks 6 and 7 of gestation by atrophy of the hyaloid artery, which runs from the optic nerve head to the posterior pole of the lens, and by closure of the lumen of the lens vesicle . Failure of resorption of this fetal vasculature leads to a vascularized membrane or stalk that may extend, completely or incompletely, from the disc to the back of the lens, denoted persistent hyperplastic primary vitreous (PHPV). Associated findings, occurring singly or in combination, are microphthalmia, glaucoma, miosis with inability to pharmacologically dilate the pupil, shallow anterior chamber caused by forward bulging of the iris, and contracted ciliary processes that are drawn in toward the pupil. The cause of this anomaly, which is not considered heritable and may be found as an isolated defect or in conjunction with other ocular malformations, is unknown. Rare cases of bilateral PHPV are commonly associated with congenital systemic malformation syndromes. Before the advent of modern diagnostic and therapeutic techniques, visual prognosis with PHPV was poor. Except in the case of retinal scarring or detachment, visual prognosis can be good with early diagnosis and prompt intervention .
Anophthalmia, microphthalmia, and colobomata of the iris and retina
With the exception of cyclopia ( Fig. 7 ), anophthalmia, or total absence of the eye, is the most severe ocular abnormality . It represents a developmental-field defect in Pfeifer’s diacephalic region, which is subject to some of the most severe craniofacial malformations, including cleft lip, palate, and nose; clefts of the Tessier-type; and anophthalmia, microphthalmia (axial length of the eye shorter than normal) ( Fig. 8 ), and cryptophthalmos (congenital absence of the eyelid). Anophthalmia, microphthalmia, and cryptophthalmos are morphogenetically related . Anophthalmia and microphthalmia are frequently found in conjunction with hypertelorism and hemifacial microsomia. They were also recently associated with congenital defects of the limb and musculoskeletal systems; other defects of the eye; defects of the ear, face, and neck; and specifically trisomy 18, trisomy 13, other chromosomal abnormalities, amnion rupture sequence, osteogenesis imperfecta, Aicardi syndrome, CHARGE syndrome, Cornelia de Lange syndrome, dwarfism, Goltz syndrome, Meckel-Gruber syndrome, Seckel syndrome, septo-optic dysplasia, short rib–polydactyly dysplasia syndrome, and Werdnig-Hoffmann disease . A large population study of the epidemiology of anophthalmia and microphthalmia in Hawaii recently reported a combined rate of 3.21 per 10,000 live births, an anophthalmia rate of 3.01 per 100,000 live births, a microphthalmia rate of 2.84 per 10,000 live births, and an association with low birth weight and earlier gestational age .
Anophthalmia, whether unilateral or bilateral, is always accompanied by hypoplasia of the associated orbits, because the eye normally stimulates orbital growth during development of the midface during childhood . Microphthalmia, if very severe, may have a similar result . If unilateral, facial asymmetry invariably results, with hypoplasia of the midface, a canted occlusal plane, and a short ascending ramus of the mandible. Therefore, especially in unilateral cases of anophthalmia and microphthalmia, orbital growth must be induced using orbital implants of progressively increasing size throughout childhood. The judicious use of self-inflating expanders, most recently of methacrylate- N -vinylpyrrolidone, has been shown to achieve a long-lasting, aesthetically pleasing, physiologic cosmetic result .
Microphthalmia may or may not be visually significant, depending on its degree. It may exist with a normal or small cornea (microcornea). Nanophthalmia, a rare form of bilateral microphthalmia, is an autosomal dominant disorder with thick sclera that predisposes patients to spontaneous retinal detachments secondary to subretinal fluid. It is commonly associated with congenital cataract (often nuclear or PHPV) and coloboma, the latter likely the primary cause of microphthalmia. Microphthalmia and coloboma may follow autosomal dominant or autosomal recessive inheritance patterns .
Coloboma, or congenital absence of a portion of the tissue of the eye, represents a failure of fusion of the embryonic fissure between the fifth and seventh week of gestation by cell division and apoptosis, leading to defects of the iris, ciliary body, choroid, retina, or optic nerve ( Fig. 9 ) . It can appear as an isolated finding or as part of a multisystem syndrome, most frequently as part of the CHARGE (Coloboma of the eye, Heart defects, Atresia of the choanae, Retarded growth and development, Genital hypoplasia, and Ear anomalies with or without deafness) syndrome and in patients who have chromosomal disorders.
Coloboma of the optic nerve may be visually insignificant, with benign enlargement of the optic disc and cup, or may involve the inferior disc with pigmentary disruption and visual loss. It may also extend into the subjacent retina, with failure of the retinal pigment epithelium (RPE) to fuse coupled with absence of the choroid in the affected portion, revealing the underlying sclera on funduscopic examination. Defective retina within the coloboma itself is prone to retinal detachment. If large, retinal coloboma may involve the macula, fovea, and optic nerve simultaneously, with a uniformly poor visual result. In some cases, coloboma may involve only the iris, resulting in a keyhole-shaped pupil of no visual significance, unless the superior pupillary edge is drawn down into the central visual axis .
Abnormalities of the optic nerve
Optic nerve hypoplasia
ONH, characterized by a small underdeveloped optic nerve ( Fig. 10 ), may be unilateral or bilateral, and may occur in isolation or with various congenital anomalies of the central nervous system (less common in unilateral cases) and pituitary axis, more commonly known as septo-optic dysplasia. When septo-optic dysplasia occurs in combination with characteristic facies, open anterior fontanelle, and other anomalies of the cerebral midline, the term de Morsier syndrome is used, although it is a common practice to use the two terms interchangeably. Mild cases of ONH may be visually insignificant and either escape detection or be seen only on funduscopic examination. More severe cases can show marked deficiency of nervehead tissue, disc pallor, abnormal vessel branching off of the disc, and a “double-ring” sign, reflecting the rim and part of the interior of the scleral canal that an optic nerve of normal size would have otherwise occupied .
Vision impairment of ONH ranges from various visual field defects and subnormal acuity to blindness, often with attendant nystagmus and strabismus. Hypothalamic or pituitary involvement in septo-optic dysplasia can cause various endocrine defects, such as growth hormone or thyroid deficiency. MRI of the brain is recommended in suspected cases to identify pituitary, hypothalamic, and cortical pathology; defects of the corpus callosum and septum pellucidum may also be identified. Endocrine evaluation is also advised. In the case of unilateral ONH, patching of the unaffected eye may help minimize amblyopia .
ONH has been associated with mutations in the HESX1 homeobox gene . Effects of fetal alcohol syndrome have been shown to result in hypoplasia of the optic nerve head in 48% of affected infants .
Morning glory disc
Morning glory disc, an almost universally unilateral condition, is characterized by the presence of an enlarged optic nerve, retinal vessels arranged in a spoke-like pattern, and a glial tuft overlying the optic cup. This anomaly may represent a coloboma of the optic disc. On CT or ultrasound, morning glory disc appears as an enlarged, funnel-shaped optic nerve as it enters the globe. Mild to severe vision impairment may be present. In some cases, patching of the unaffected eye may be useful to minimize amblyopia. In rare cases, morning glory anomaly may signify a basal encephalocele, especially with a concomitant notch of the central lower lip .
Normal optic nerve variants
Several normal variant optic nerve configurations exist, none of which is associated with decreased vision in the affected eyes. One anomaly involves peripapillary pigmentation, in which the head of the optic nerve is surrounded by a ring of hyperpigmented tissue. When this defect exists on the disc’s temporal side, as is more commonly the case in myopia, it is referred to as a temporal crescent ( Fig. 11 ). Peripapillary hypopigmentation may represent an area of retinochoroidal atrophy. Another developmental variant involves tilting of the optic disc, most commonly, but not necessarily, secondary to the increased globe length seen in myopia. In these cases, the disc appears slanted on funduscopic examination, with the nasal portion of the nerve more prominent than the temporal, which may make assessment of the optic nerve cup size difficult.
A third variant commonly seen in childhood is physiologic cupping of the optic nerve. This defect is most commonly bilateral and symmetric, but may be unilateral or asymmetric and is believed to have autosomal dominant inheritance when the parents are affected. The cup/disc ratio may be as high as 0.8, and the physiologic cup will typically display very sharp edges with some splaying of the vessels as they exit the cup. Glaucoma must be ruled out in a child who presents with a high cup/disc ratio, and referral to an ophthalmologist is advised.
A fourth variant, most commonly seen in hyperopic children, is pseudopapilledema, in which the optic nerve head appears swollen although no true edema or evidence of increased intracranial pressure is present. The edges of the disc may be blurry and the disc may appear elevated, but other features of true papilledema, such as disc hyperemia, absence of spontaneous venous pulsations, and small hemorrhages or exudates of the nerve fiber layer, will be absent. Pseudopapilledema may be the result of buried drusen, which may be identifiable only with imaging studies such as CT or ultrasound. Rarely, these may be associated with visual field defects or subretinal fluid leakage .
Congenital abnormalities and syndromes of the eye
Abnormalities of the anterior segment of the eye
Anterior segment dysgenesis
A significant number of congenital ocular anomalies, all of them rare, involve the anterior segment of the eye, which includes the cornea, iris, ciliary body, and lens. The most common of these uncommon disorders is termed the Axenfeld-Rieger spectrum of anomalies , which includes several abnormalities, such as posterior embryotoxon (an arcuate white line in the periphery of the cornea), abnormalities of pupillary shape or location, polycoria (full-thickness defects of the iris independent of pupillary defects), and adhesions between the iris and cornea. These ocular abnormalities may coexist with a spectrum of extraocular anomalies as part of an autosomal dominant multisystem congenital disorder that includes facial dysmorphism, redundant periumbilical skin, and anomalies of dentition. Many patients who have Axenfeld-Rieger develop glaucoma, and therefore should be periodically screened for this condition .
Peter’s anomaly
Children born with a characteristic white opacity of the cornea are classified as having Peter’s anomaly. The opacity is usually, but not necessarily, an avascular, central defect that causes a significant visual disturbance. Patients often also exhibit iridocorneal adhesions with or without an anteriorly displaced cataractous lens. Peter’s anomaly may be unilateral or bilateral, and patients are predisposed to glaucoma. In the absence of absolute contraindications, surgery for Peter’s anomaly, which includes corneal transplantation with or without lens extraction, must proceed at an early age to avoid irreversible amblyopia resulting from visual deprivation in the first weeks of life. An alternative procedure involves the resection of a large section of iris (sector iridectomy) to allow the child to see around the corneal opacity .
Peter’s anomaly is known to occur in isolation, as part of a larger ocular syndrome such as aniridia or microphthalmia, or as part of a systemic syndromic presentation in Peter’s Plus syndrome, involving developmental delay, short stature, and skeletal dysplasia .
Peter’s anomaly is believed to represent a nonheritable disorder and has been associated with mutations in the PAX6 gene and the CYP1B1 gene for congenital glaucoma .
Other congenital opacities of the cornea
Several other corneal opacities are congenital in origin. A primary bilateral, gray-white corneal opacification is seen in congenital hereditary endothelial dystrophy (CHED) involving the innermost corneal layer and in congenital hereditary stromal dystrophy (CHSD) involving the middle layer of the cornea. A primary white vascularized opacification of the cornea is characteristic of sclerocornea; focal white opaque lesions suggest corneal dermoid . Although not typically threatening to the visual axis, dermoids of the corneal limbus, which are small white masses that often have hairs growing from them, may cause amblyopia secondary to astigmatism, may cause ocular discomfort, and are seen in Goldenhar syndrome (oculo–auricular–vertebral spectrum). Goldenhar syndrome results from aberrant development of the first or second branchial arch and includes, in addition to epibulbar dermoids, hemifacial microsomia with resultant orbital asymmetry, small ears with skin tags, and vertebral anomalies .
Corneal clouding may also occur secondary to a host of metabolic storage diseases (although the cornea is often clear at birth), cystinosis, congenital infection, and birth or amniocentesis trauma. Forceps injury to the cornea with rupture of Descemet’s membrane may result in corneal clouding caused by edema. A large-appearing, cloudy cornea is a frequent presentation of congenital glaucoma ( Fig. 3 ) .



