Congenital and Neonatal Corneal Abnormalities
Matthew K. Perez
Sadeer B. Hannush
George O. Waring III
This chapter presents a summary of congenital and neonatal corneal abnormalities that occur within the first few months of life. Although these conditions are rarely encountered by the general ophthalmologist, an accurate diagnosis is crucial, given the possibility of permanent blindness. Corneal abnormalities contribute to approximately 1% of childhood blindness in Europe and North American and a higher percentage in Africa and South Asia.1,2 Workup must also include identification of associated ocular and systemic disease that often accompany neonatal corneal abnormalities. When appropriate, genetic counseling should be sought and surgical or medical therapy initiated.
EXAMINATION OF THE CORNEA IN AN INFANT
A thorough history is the first step in evaluation of an infant with a corneal abnormality. This should include medical records for the child and a detailed obstetrical history. Any maternal exposure to drugs, toxins, or infectious agents during pregnancy, as well as any medications prescribed for the infant should be noted. It must also be determined if there is any family history of congenital disease.
There will be some instances in which an examination under anesthesia is required; however, this should be used as a last resort given the cost in added time and use of an operating suite. One useful method for an infant ocular examination is to instruct the parents to keep their child hungry before the visit. This will allow feeding to serve as a distraction and assist in keeping the child quiet during the examination. Another useful technique in maintaining comfort for the infant is to perform as much of the examination as possible with the child in the parent’s lap, having the parent handle and position the child when needed. Once calm and in position, instill topical anesthetic and place either a lid speculum or Koeppe lens to begin the examination (Fig. 1A). Instruments used in the evaluation of neonatal corneal abnormalities are listed in Table 1.
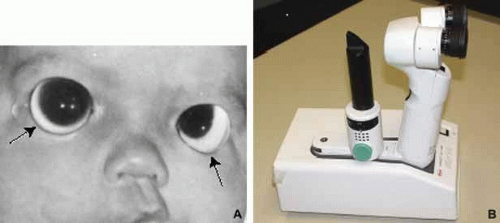 Fig. 1 Instruments used during an infant ocular examination. A. A Koeppe lens is used to allow direct visualization of the angle. (courtesy of Donelson Manley, MD) B. Portable slit lamp biomicroscopy permits a detailed anterior segment examination with zoom optics and the capability for photography. (courtesy of Nicholas Pefkaros, MD) |
TABLE 1. Evaluation of Neonatal Corneal Abnormality | |||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||
Portable slit lamp biomicroscopy is a useful technique for evaluation of the anterior segment (Fig. 1B). It is preferable to have a model with zoom optics and the capability of photography like the Kowa II portable slit lamp. This is optimal for obtaining good pictures and making the most accurate drawings of any abnormalities.3 Gonioscopy can then be performed with the Koeppe lens in place, but the lens must be removed for tonometry. If the cornea is heavily scarred or the globe is grossly distorted, then intraocular pressure must be estimated by palpation. After examination of the anterior segment, fundoscopy or other indicated studies (ERG, ultrasonography) can be performed. In some cases, severe corneal opacification will prevent visualization behind the cornea, and ultrasound biomicroscopy should be used to detect other anterior segment or vitreoretinal abnormalities.4 A complete infant ocular examination can be performed in approximately 2 hours.
ABSENCE OF THE CORNEA AND CRYPTOPHTHALMOS
Agenesis of the cornea is an extremely rare condition resulting from failure of the anterior segment to differentiate despite formation of the optic cup.5 The eye becomes completely surrounded by a scleral-like band of fibrous tissue. Absence of the cornea does not occur in isolation, rather it is always accompanied by the absence of other anterior segment structures such as the iris and ciliary body. Clinically, this condition can appear as a form of microphthalmos or apparent anophthalmos.
Cryptophthalmos is another relatively rare condition characterized by replacement of the eyelids with facial skin covering the orbit from the forehead to the cheek, connecting to the underlying globe and obliterating the cornea (Fig. 2A).6 This condition represents metaplastic change of the corneal epithelium and conjunctiva into stratified squamous keratinized epithelium.7 Cryptophthalmos can be unilateral or bilateral and usually occurs sporadically along with other congenital malformations including dyscephaly, syndactyly, and urogenital malformations. Although the pathogenesis is unclear, animal models suggest possible causes of cryptophthalmos syndrome to include defects in programmed cell death or vitamin A metabolism.8 An autosomal-recessive pattern of inheritance has been described for cryptophthalmos in as many as 25% of cases examined.9 Recent reviews have detailed the association of cryptophthalmos with other congenital malformations.8,9
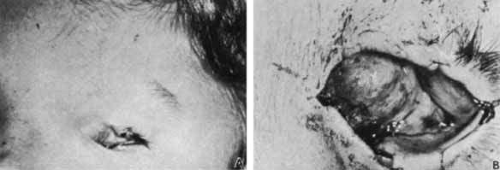 Fig. 2 Cryptophthalmos. A. Patient with partial cryptophthalmos demonstrates skin adherent to the cornea, lack of eyelids medially, and abnormal eyelids laterally. Hair stream extends across forehead to cryptophthalmic area. B. Postoperative appearance shows dissection of the skin from the cornea with attachment to the lateral eyelids. The cornea is thin and covered with fibrovascular tissue. (Waring GO, Shields JA: Partial unilateral Cryptophthalmos with syndactyly, brachycephaly, and renal anomalies. Am J Ophthalmol 79:437, 1975) |
True (complete) cryptophthalmos, the most common form of this entity, is the result of failed formation of eyelid folds such that lashes and eyebrows are absent. In addition, lacrimal glands and canaliculi are generally absent. Although palpable, the underlying globe is microphthalmic with anterior chamber malformations. B-scan ultrasonography shows in a majority of cases the replacement of the remainder of the cornea and anterior chamber with connective tissue. Partial cryptophthalmos involves only the medial aspect of the eyelid, leaving the lateral eyelid normal in structure and function. Another form is abortive cryptophthalmos, or congenital symblepharon, in which only the upper eyelid undergoes metaplasia and remains adherent to the upper third of the cornea.10
Histopathologically, the skin consists of stratified squamous keratinized epithelium and an underlying dermis without appendages, which is fused to the fibrovascular tissue replacing the cornea. The anterior chamber is small or nonexistent, and the trabecular meshwork, Schlemm’s canal, iris, and lens are absent.11 Surgical correction has been attempted for both complete and partial cryptophthalmos. The major goals of surgical intervention are to achieve a cosmetic improvement, to preserve the globe, and to protect any remnant of the cornea from infection (Fig. 2B).12 There has been some success in restoring good visual function in patients with partial cryptophthalmos; however, this is not the true for cases of complete cryptophthalmos.12
MEGALOCORNEA
The horizontal diameter of the newborn cornea is usually 10 mm, reaching the normal adult size of 11.8 mm by 2 years of age.13 Megalocornea is a congenital condition defined by a horizontal corneal diameter of greater than 12 mm in the newborn or greater than 13 mm in an adult without an associated elevation in intraocular pressure.14 Most often this is a nonprogressive bilateral symmetric corneal enlargement. There are three basic patterns of megalocornea (Table 2):
TABLE 2. Differential Diagnosis of Enlarged Cornea | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Simple megalocornea, which is unassociated with other abnormalities.
Anterior megalophthalmos, in which the ciliary ring and lens are enlarged.
Buphthalmos in association with infantile glaucoma.
A-scan ultrasonography can be used to distinguish simple megalocornea from anterior megalophthalmos and buphthalmos. Only the anterior chamber will be enlarged in anterior megalophthalmos, whereas the entire globe will be enlarged in buphthalmos. Keratoglobus is a condition in which there is generalized thinning of the cornea, which may appear to be enlarged clinically but is usually normal in diameter.
SIMPLE MEGALOCORNEA
These individuals present with bilateral clear corneas of normal thickness measuring greater than 12.5 mm in diameter. This condition is not progressive and there are no associated ocular or developmental abnormalities.15 In the few pedigrees reported, this condition may possess autosomal-dominant inheritance.16 Management is similar to anterior megalophthalmos discussed later. There is no treatment for this condition except for the correction of any refractive error and reassurance to the family that eye can function normally. Histopathologically, the anterior chamber is normal except for an enlarged cornea, and there are no abnormalities of the posterior segment.15
ANTERIOR MEGALOPHTHALMOS
Megalocornea most commonly presents as anterior megalophthalmos, in which there is abnormal development of other anterior segment structures in addition to the cornea (Fig. 3).17 The cornea is clear with normal curvature and thickness; however, some affected corneas may develop a central mosaic stromal opacity.18 The anterior chamber appears deep from either the steep arc of the enlarged cornea or secondary to lens subluxation and iridodonesis. Other notable features of anterior megalophthalmos include anterior iris stromal hypoplasia, peripheral full-thickness iris holes, and Krukenberg’s spindle and pigment dispersion with iris transillumination defects (Fig. 3B). Dilator muscle atrophy can lead to miotic pupils.17
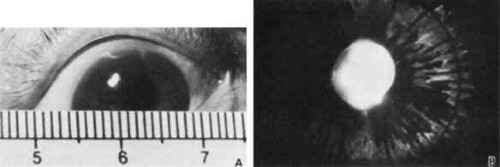 Fig. 3 Anterior megalophthalmos. A. Megalocornea with horizontal diameter of 14 mm. B. Iris transillumination shows defects in the iris pigment epithelium. Patient had cataract extraction at age 37. |
Systemic disease is occasionally identified in patients with anterior megalophthalmos and may include neurological, skeletal, and dermatological abnormalities.19,20 Anterior megalophthalmos is most commonly inherited in an X-linked recessive fashion, although autosomal-dominant and autosomal-recessive patterns of inheritance have been described.5
Although the pathogenesis remains unclear, it is generally accepted that this condition is the result of disturbed growth and development of the neural crest cells that comprise the anterior chamber. Developmentally, megalocornea can result from incomplete growth of the optic cup anteriorly, leading to an enlarged cornea occupying this extra space.21 This may explain the occurrence of megalocornea in association with other anterior chamber abnormalities, including Rieger’s syndrome (e.g., prominent Schwalbe’s ring, iris strands to Schwalbe’s ring, hypoplasia of anterior iris stroma) and congenital glaucoma with buphthalmos.22 Linkage studies have demonstrated a possible gene locus in the region Xq13-q2523, and identification of specific genes at this locus may have future implications in understanding the pathogenesis of megalocornea.24
Patients with megalocornea need to be followed-up regularly throughout their life with annual examinations to evaluate for lens subluxation, premature cataract development, and elevations in intraocular pressure. These are the major complications of megalocornea for which treatment is available and restoration of vision is possible. Cataract extraction in patients with megalocornea has a higher risk of complications because of lens dislocation. Vitreous loss may require an anterior vitrectomy in some cases. Implantation of an intraocular lens may also be difficult, sometimes requiring placement of an iris supported or sutured lens.25 Recent studies, however, suggest that capsulorhexis with placement of a standard intraocular lens, move as indicated in comments with the use of a capsular tension ring, can be safely performed without complication.26 Management of elevated intraocular pressure is similar to that for treatment of chronic open-angle glaucoma.
BUPHTHALMOS
Buphthalmos is an enlargement of the entire globe, including the cornea, secondary to elevated intraocular pressure in infantile glaucoma. Although not fitting the classic definition of megalocornea, buphthalmos is worth mentioning because it is part of the differential diagnosis of an enlarged cornea. This diagnosis must be considered in any patient presenting with an enlarged cornea. Prompt identification and treatment of infantile glaucoma will save a child’s vision. This will be covered in more detail in Volume 3, Chapter 19 of this series.
KERATOGLOBUS
Keratoglobus is a rare bilateral condition in which there is generalized thinning and globular anterior protrusion of the cornea (Fig. 4A). The iris and lens remain unaffected. In some cases the cornea can become enlarged, confusing this diagnosis with megalocornea.27 Various connective tissue abnormalities are associated with keratoglobus, including hypermobile joints and a thinned sclera, which may appear blue from the underlying uveal tissue.28 Specific connective tissue diseases that may present with keratoglobus include osteogenesis imperfecta or Ehlers Danlos syndrome type VIA (Fig. 4B).29 Keratoglobus has also been found in association with autosomal-recessive Leber congenital amaurosis30 and pellucid marginal corneal degeneration.31 Although usually present at birth, keratoglobus may not be recognized until childhood when the signs of connective tissue disease or blue sclera become apparent. Despite most cases being congenital, adult-onset keratoglobus has been found associated with thyroid ophthalmopathy32 and vernal conjunctivitis.33
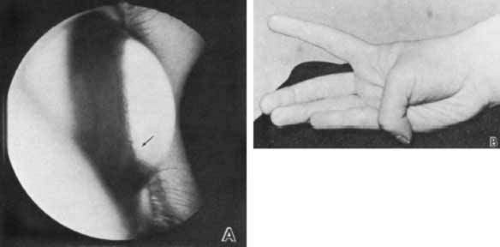 Fig. 4 Keratoglobus. A. Lateral view demonstrates anterior protrusion of thin cornea. Iris process in the angle are visible (arrow) without gonioscopic lens. B. Increased flexion of thumb emphasizes association with lax joints. Keratoglobus is sometimes classified as Ehlers-Danlos syndrome type VIA. |
The steep arc of the cornea results in a deep anterior chamber and an average keratometry reading of greater than 55 diopters.34 A faint central stromal haze may be present, but the stress lines and subepithelial scarring characteristic of keratoconus do not occur. Histopathological analysis demonstrates a disorganized stromal architecture34 along with stromal thinning.28,35 Focal disruptions occurring in Bowman’s layer may produce the faint opacity seen clinically. Acute corneal edema may occur from spontaneous breaks in Descemet’s membrane. This acute hydrops will resolve in time and requires no treatment.
The thin and disorganized stroma results in a cornea that is very susceptible to perforation after minor trauma.29 It is imperative that parents are counseled regarding the need for a safe environment and the mandatory use of protective glasses or eye guards when necessary.
To prevent the development of amblyopia, early diagnosis and treatment is essential. Initial management of refractive error with spectacles or contact lenses may be sufficient. Surgical intervention is required in some cases of advanced keratoglobus or in the event of a ruptured globe after minor trauma.36 Various surgical techniques have been used and are complicated by the thin and irregular cornea and sclera.34,37,38 Newer surgical approaches are being developed to reduce graft failure and improve visual outcome.39,40
MICROCORNEA
Microcornea is defined by a horizontal corneal diameter of less than 11 mm.14 In this condition, the corneal diameter is usually between 7 and 10 mm, with some being reported as small as 4 mm.41 Accurate determination of the corneal diameter may be complicated in some cases by scleralization of the corneal limbus. The cornea is usually clear with normal thickness, and its shortened diameter usually creates a steep curvature with an increased refractive power. Instances of a flattened cornea with keratometry readings of less than 35 diopters have been reported in severe microcornea42 or in microcornea with sclerocornea.43
The wide variety of ocular and systemic abnormalities that accompany microcornea make generalizations about this condition difficult. Associated ocular features can include other anterior segment malformations, such as sclerocornea or aniridia.43 Microcornea can also present in cases of nanophthalmos, also called simple microphthalmos, in which the primary abnormality is a total reduction in the size of the globe.44 Microcornea may also occur with congenital microphthalmos, in which there are multiple ocular abnormalities in conjunction with a small globe (Fig. 5).45 Isolated cases of microcornea have also been reported.46 Microcornea can be a sporadic condition, or it can be inherited in either autosomal dominant or autosomal-recessive patterns.47
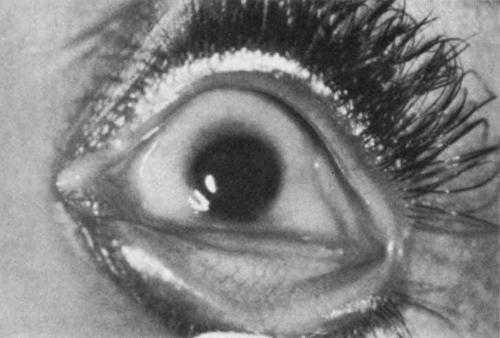 Fig. 5 Microcornea. This 6-mm-diameter cornea was part of congenital microphthalmos. |
Assessment with A-scan ultrasonography is an important study in all patients with microcornea to establish if it is the cornea alone, or if the entire globe is small. Transillumination of the ciliary ring may help provide an accurate measurement of the true corneal diameter in cases with scleralization of the limbus. This will help distinguish microcornea from sclerocornea or cornea plana, in which an indistinct limbus produces the clinical appearance of a small cornea.
One histopathologic study of microcornea demonstrated a transformation of deeper corneal layers into scleral tissue at the true limbus.48 The scleral spur and Schlemm’s canal were absent, with thinning and atrophy of the ciliary muscle. No changes were found in Descemet’s membrane.
Management of microcornea must include careful examination looking for other ocular conditions. Cataract must be ruled out because one presentation is the microcornea–cataract syndrome.43 Routine examinations must also include intraocular pressure measurements to rule out glaucoma, which can occur in up to 20% of all cases.49 In isolated microcornea, which can present as either myopia or hyperopia, correction of refractive error can provide an improved visual outcome.
NEONATAL CORNEAL OPACITIES
An infant with a corneal opacification presents a diagnostic challenge for any ophthalmologist. A useful acronym to aid in recalling the differential diagnosis is STUMPED (Table 3). This mnemonic will hopefully prevent an ophthalmologist from becoming stumped when facing these conditions. The incidence of the different types of neonatal corneal opacities is unknown, but Peters anomaly and infantile glaucoma are the most common.
TABLE 3. Differential Diagnosis of a Neonate with Cloudy Cornea (STUMPED) | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
SCLEROCORNEA (STUMPED)
Sclerocornea is a condition in which normal cornea is replaced by white, vascularized tissue resembling the sclera, obliterating the limbus and scleral sulcus, and leaving a clearer central cornea (Fig. 6). This nonprogressive congenital anomaly is thought to result from defective mesenchymal migration at an early stage of differentiation.50 Given the broad use of the term sclerocornea in a wide variety of corneal disease, it is not considered a distinct diagnostic entity.51
 Fig. 6 In peripheral sclerocornea, the peripheral white corneal opacity blends with the sclera. This obliterates the limbus and scleral sulcus, leaving the central cornea clearer than the periphery. |
Both sexes are affected by sclerocornea and a majority of cases have bilateral involvement, which may be asymmetric. Half of all cases are sporadic, with the rest having either dominant or recessive patterns of inheritance,51,52 with recessive forms usually more severe than dominant forms.53 A number of ocular and systemic abnormalities are associated with sclerocornea, including skeletal and central nervous system abnormalities.54 Chromosomal abnormalities may also occur.53,55
Clinical presentation of sclerocornea can be quite varied. In some forms only a 1- to 2-mm rim of peripheral cornea is involved, leaving the central cornea clearer with distinct margins. More involved cases can include nodular extensions of scleral tissue into more central portions of the cornea. Sclerocornea can also present as a diffusely opaque cornea with peripheral vascularization or as a completely white and vascularized cornea. The fine, superficial vasculature appears to originate from the conjunctival vasculature.51 Either penlight or scleral scatter with the slit lamp best reveals the extent of sclerocornea. Diagnosis can be complicated in cases in which the entire cornea is involved and visualization of the anterior segment is limited. Ultrasound biomicroscopy is useful in assessing these corneas and establishing a diagnosis.56
Sclerocornea is classified into four groups, although distinction among them is often imprecise: (1) isolated sclerocornea; (2) sclerocornea plana (cornea plana); (3) peripheral sclerocornea with anterior chamber cleavage abnormalities; and (4) total sclerocornea.
ISOLATED SCLEROCORNEA
SCLEROCORNEA PLANA
Sclerocornea plana is characterized by scleralization and flattening of the corneal surface with keratometry readings less than 38 diopters. This condition is bilateral and can be either sporadic or inherited as an autosomal-dominant trait.59 Sclerocornea plana has been reported in association with epidermolysis bullosa dystrophica and spontaneously reabsorbed congenital cataracts.60 Although vision may be normal, amblyopia with strabismus, aniridia with cataract, flattening of the anterior chamber, and infantile glaucoma sometimes occur. Patients may develop pseudoptosis because of poor upper lid support from the flattened cornea. In sclerocornea plana, the peripheral rim of the cornea is more involved, leaving a clear central cornea with normal thickness. The blending of the peripheral cornea with the surrounding sclera makes accurate measurement of the corneal diameter difficult. Refractive error may range from moderate hyperopia to moderate myopia.
PERIPHERAL SCLEROCORNEA WITH ANTERIOR CHAMBER CLEAVAGE ABNORMALITIES
Peripheral sclerocornea presents with an indistinct corneoscleral limbus as described, and this is often a common feature in both Reiger’s syndrome and Peters anomaly.61 In cases in which the entire cornea is opaque, distinction of sclerocornea, with its slightly clearer central zone, from Peters anomaly, which has a central corneal opacity, becomes difficult. One distinction is the presence of iridocorneal adhesions in Peters anomaly. Histologically, this distinction between Peters and sclerocornea can also become complicated. Some patients with clinically apparent sclerocornea may have focal loss of the corneal endothelium and Descemet’s membrane, findings more typical of Peters anomly.50 Conversely, some eyes with Peters anomaly are found histologically to have a thin, yet intact, Descemet’s membrane.62 In these circumstances, a precise discrimination between these two conditions is not necessary because their management is similar.
TOTAL SCLEROCORNEA
In cases in which the entire cornea is completely white and vascularized, an accurate diagnosis becomes more difficult (Fig. 7). Ultrasound biomicroscopy should be used before penetrating keratoplasty to define other underlying anterior segment abnormalities, such as corneolenticular adhesions, which may be present.56,63
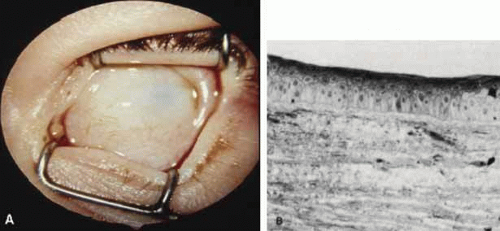 Fig. 7 Sclerocornea. A. Total sclerocornea manifests porcelain white cornea with superficial arborizing vessels. B. The irregular superficial stroma contains scattered blood vessels (Toluidine blue, ×350). |
Histopathologically, the epithelial thickness is variable, with rete peg-like fingers penetrating a fragmented or absent Bowman’s layer. The epithelial basement membrane remains intact. The anterior stromal architecture is disorganized with fibrils of collagenous tissue 70 to 150 nm in diameter (normally 25 to 30 nm).58 The posterior stroma may maintain its normal lamellar architecture. This random pattern of fibrils, along with the presence of elastic fibers and blood vessels, gives the cornea a sclera-like appearance. Descemet’s membrane is abnormal, appearing either as a thin irregular layer with collagenous tissue behind it or as having focal dehiscences that may contain fibrous tissue.57
This condition should not be confused with Mieten’s syndrome, which includes superficial vascularized corneal opacities that resemble sclerocornea. Other associated features are mental retardation, growth failure, and abnormally short ulnae and radii.
TEARS IN THE ENDOTHELIUM AND DESCEMET’S MEMBRANE (STUMPED)
Tears or breaks in the endothelium and in Descemet’s membrane can be caused by either birth trauma or infantile glaucoma. Because the adult structure of the cornea is not reached until approximately 6 months of age,64 the cornea of an infant is more elastic and prone to injury secondary to elevations in intraocular pressure. The elevated pressure produced either acutely by birth trauma or chronically in infantile glaucoma can distend the infant’s globe, exceeding the elasticity of Descemet’s membrane, resulting in tears. In either circumstance, the tears or breaks allow aqueous to enter the stroma and epithelium, producing corneal edema. The disparate clinical settings for birth trauma and infantile glaucoma should produce little confusion as to the cause of the tear.
Acute ocular birth trauma is being seen less often by ophthalmologists because of improved prenatal care and elective cesarian section reducing the rate of complicated forceps deliveries (Fig. 8A). Ocular trauma from forceps is thought to occur when the forceps blade slides over the inferior orbital rim, compressing the globe against the superior orbit and stretching the cornea horizontally.65 Because the cornea is being compressed horizontally, these tears will line up in a vertical or oblique pattern (Fig. 8B). Other clinical findings associated with ocular birth trauma include ipsilateral periorbital edema and ecchymosis.
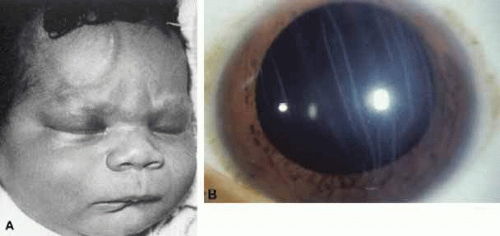 Fig. 8 Tears in Descemet’s membrane. A. Ocular birth trauma from a complicated forceps delivery. Periorbital edema and ecchymoses accompany the trauma. (courtesy of Donelson Manley, MD) B. Birth trauma produced these oblique tears in Descemet’s membrane. The edges of each break form a prominent refractile ridge that protrudes from the posterior cornea. |
Corneal edema is the most common presenting sign of infantile glaucoma when it presents in the first 5 days of life, being present in 80% of cases.66 By 6 months of age, approximately 75% of affected infants will manifest the corneal enlargement and buphthalmos characteristic of infantile glaucoma.66 The tears in Descemet’s membrane caused by infantile glaucoma have a more random distribution, often circumferential to the limbus and sometimes extending sinuously toward the center of the cornea. Clinical features such as a delay in presentation of corneal edema, patterns of Descemet’s tears, and associated ocular findings help to clearly distinguish ocular birth trauma from infantile glaucoma.
When Descemet’s membrane tears, it retracts more in the center leaving a dehiscence with roughly parallel edges.67 The stroma and epithelium overlying this tear will become diffusely edematous. Along the margin of the tear, Descemet’s membrane will often curl toward the stroma like a watch spring. Clinically, this curl forms a refractile edge evident under retroillumination through the corneal edema. It is important not to mistake these prominent edges for the tear itself, which is actually the space between these edges. To perform a complete anterior segment evaluation, it may be necessary to remove the edematous epithelium with a moist cotton swab. The corneal edema should resolve within weeks to months, provided the birth trauma was not too severe and that the intraocular pressure was surgically lowered in cases of infantile glaucoma.
With time, a fresh endothelium will resurface the posterior cornea and synthesize a new basement membrane, filling the dehiscence and accentuating the edges of the tear. After the corneal edema resolves, the edges of the break will appear as rounded, glassy ridges protruding from the posterior cornea readily apparent under retroillumination. In some instances, one edge of the tear may separate from the stroma and hang into the anterior chamber as a falciform ledge, with its free edge curling anterior to form a scroll (Fig. 9). When two tears occur parallel to each other, the edge of Descemet’s membrane between the tears may curl toward each other. This strip of Descemet’s may then disassociate from the overlying stroma, resulting in a glassy strand across the concavity of the posterior cornea. In this circumstance, the newly laid basement membrane will have a beaten-metal, guttate appearance.
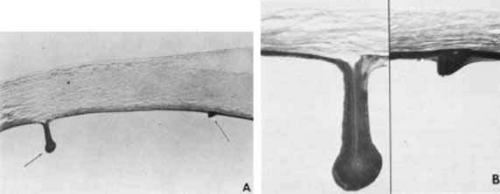 Fig. 9 Histopathology of healed tears in Descemet’s membrane from congenital glaucoma. A. Regenerating corneal endothelium has produced new basement membrane in the bed of the tear and over each edge (arrows) of the tear. B. Main figure demonstrates one edge of the tear in Descemet’s membrane, which has separated from the overlying stroma and coiled anteriorly like a watch spring. Regenerated basement membrane has encased this and formed a prominent ledge. Inset. Other edge of the tear, in which regenerated basement membrane forms a ridge protruding into the anterior chamber. (Waring GO, Laibson PR, Rodrigues MM: Clinical and pathologic alterations of Descemet’s membrane: With emphasis on endothelial metaplasia. Surv Ophthalmol 18:325, 1974) |
Histopathologically, the edge of a Descemet tear curls toward the stroma, possibly because of the differential elasticity between the anterior banded and posterior nonbanded layers within Descemet’s membrane.67 As the new regenerating endothelium spreads over the edge, it encases the original coiled Descemet’s membrane in a thick multilaminar periodic acid-Schiff (PAS)-positive basement membrane (Fig. 9). This new basement membrane forms the clinically evident refractile edge. Within the bed of the tear, the regenerating endothelium lays down an irregular basement membrane with focal excrescences that produce the beaten-metal appearance.
Ultrastructural studies of the cornea in human congenital glaucoma are rare,68 but the ultrastructure in rabbits with spontaneous buphthalmos has been described in detail.69 Intraepithelial and subepithelial edema is present, accounting for the ease with which one can remove the epithelium clinically. Stromal edema, loss of regular collagen architecture, and the degree of scarring correlate with the degree of corneal opacity and thickness. In rabbits with mild to moderate corneal clouding, Descemet’s membrane is thicker than normal and manifests the compact structure typical of basement membrane. In rabbits with more severe corneal opacification, a layer of small-diameter collagen fibrils enmeshed in amorphous material appears posterior to Descemet’s membrane. In these rabbits, no tears in Descemet’s membrane are observed clinically or histopathologically, suggesting that corneal edema may occur from elevated intraocular pressure alone. The thin endothelial cells can be as large as four times their normal size and contain cytoplasmic vacuoles appearing as small pits on scanning electron microscopy. In humans, these enlarged endothelial cells may represent the ones that decompensate later to produce corneal edema. Recently, in vivo confocal microscopy has been used to further evaluate Descemet’s tears and endothelial changes associated with congenital glaucoma.70 Key findings include a mild reduction of keratocyte density in the middle and posterior stroma, along with the presence of discontinuous hyperreflective structures overhanging the endothelial layer at the level of Descemet’s membrane. Additional confocal analysis of the endothelial layer demonstrate a low cell count, focal cellular lesions, severe polymegethism, and pleomorphism.70
Treatment is generally not necessary for patients with ocular forceps injury. The corneal edema will usually resolve within weeks. Corneal astigmatism may result from this injury and will require early refraction and patching to prevent amblypoia.65 In some instances, the edema may persist and form an opacification, which may require penetrating keratoplasty. In this circumstance, the visual prognosis is poor in the affected eye. Young children with infantile glaucoma may require surgery to control intraocular pressure. Treatment of infantile glaucoma may result in a decrease in buphthalmos and a change in corneal diameter toward normal; however, some residual corneal edema may persist. Under these circumstances, early penetrating keratoplasty is indicated and can be successful.71 Decompensation of previously stressed endothelium from either infantile glaucoma or birth trauma can occur 20 to 30 years after the original insult, requiring penetrating keratoplasty to restore vision.72 In one study of endothelial decompensation related to infantile glaucoma, the overall success rate for penetrating keratoplasty was 75%; with adequate control of intraocular pressure being a key factor in preventing graft failure.73
Another form of prenatal trauma that can produce corneal opacification is an amniotic band, which by stretching across the face during development may cause a cleft lip occasionally associated with microphthalmia and a hazy cornea. This corneal haze is likely the result of direct trauma to the cornea and not from tears in Descemet’s membrane.74
CORNEAL ULCERS AND INFLAMMATION (STUMPED)
Corneal ulcers are rarely present at birth, but when an epithelial defect does occur in a neonate, the differential diagnosis should include herpes simplex keratitis, bacterial keratitis, and neurotrophic keratitis.
HERPES SIMPLEX KERATITIS
Ocular involvement occurs in 10% to 40% of neonates with herpes simplex infection and usually appears 1 to 5 months postpartum as a purulent conjunctivitis with a dendriform or geographic epithelial defect (Fig. 10). Eyelids are usually edematous and erythematous and may or may not have vesicular lesions. Ocular manifestations include optic neuritis, cataracts, or chorioretinitis, the latter being a late manifestation. Infection usually occurs during vaginal delivery; however, cases of acquisition in utero have been documented,75 and up to 10% can occur as postnatal infections.76 In studies in which the ocular virus was typed, 15% to 30% were HSV type I.75,77 Topical povidone–iodine is an effective prophylaxis for herpes simplex, as well as for other viruses, fungi and bacteria.78 Topical silver nitrate prophylaxis is not effective.79
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


