Chapter 137 Combined Hamartoma of the Retinal Pigment Epithelium and Retina
Combined hamartomas of the retina and retinal pigment epithelium (CHRRPE) are benign tumors that may cause significant visual loss depending upon location. Accurate diagnosis is essential because the lesion may resemble a choroidal melanoma or other intraocular tumor. Combined hamartomas are usually solitary, unilateral lesions located at the optic disc or posterior pole. They typically appear slightly elevated and have varying amounts of pigmentation, vascular tortuosity, and epiretinal membrane formation. A hamartoma is defined as a benign overgrowth of cells that are normally present. CHRRPE lesions are characterized by predominant tissue subtype, including pigment epithelial, vascular or glial.1
Historical review
Early reports on combined hamartomas describe lesions that were clinically mistaken for choroidal malignancy. Histopathologically, these lesions were described as hyperplastic retinal pigment epithelium and were located juxtapapillary, in the posterior pole or peripherally.2,3 In 1961, the term “hamartoma” was used to describe a large lesion of the posterior pole in a child.4 In 1973, Gass5 reported lesions in five children and two young adults, and used the term “combined hamartoma.” In 1984, Schachat et al.,1 described 60 patients with combined hamartomas in a collaborative effort of the Macula Society Research Committee. Gass6 reported an additional 35 patients in his discussion of the Macula Society’s report. Additionally, there have been more recent reports of combined hamartomas, including a review of visual acuity outcomes of 77 patients from a single institution.7
Epidemiology
In the Macula Society’s report1 of 60 patients with combined hamartomas, the mean age at the time of diagnosis was 15 years, with a range of 10 months to 66 years. The number of males and females was equal, and three patients were black. In a recent report by Shields et al.7 which evaluated 77 patients with CHRRPE, the mean age of diagnosis was 11.9 months with a range of 0.4–60 months. There is no apparent predilection based on sex or race.
Clinical manifestations
Symptoms
Reviews of several large reports show that painless visual loss is the chief complaint in the majority of patients followed by strabismus, floaters, leukocoria and ocular pain. Detection during routine examination was reported in 10% of patients.1,7,8
Visual acuity
Visual function varies with the location of the lesion.7 Direct macular involvement including the optic nerve, papillomacular bundle, or fovea may reduce visual acuity. Extramacular lesions may cause visual loss from indirect macular distortion related to tractional forces. Other secondary causes of visual loss include choroidal neovascularization,1,9 vitreous hemorrhage,10,11 exudative retinal detachments,1 retinoschisis,1,12 and macular hole formation,13 although these are uncommon. In the Macula Society report,1 visual acuity was 20/40 or better in 45% of patients, and it was 20/200 or worse in 40%. In a review by Font et al.,8 28% were 20/40 or better, whereas 28% were worse than 20/200. A report by Shields et al.7 showed age, symptoms and visual acuity were related to tumor location. Compared with extramacular lesions, macular lesions were associated with younger presentation (mean age 14.2 months versus 9.5 months), strabismus (25 versus 31%) and decreased visual acuity (37% versus 43%) and visual acuity ≤20/200 (25% versus 69%). Mean visual acuity of macular and extramacular CHRRPE lesions was 20/320 and 20/80, respectively.
Ophthalmoscopic appearance
Combined hamartomas may be located on the optic disc and juxtapapillary region, in the macula, and in the mid-periphery. The ophthalmoscopic appearance varies depending on the location of the tumor. Lesions of the posterior pole have varying amounts of retinal pigment epithelial, vascular, and glial components, and one tumor type tends to predominate (Fig. 137.1). Clinical features include an elevated pigmented mass involving the RPE, retina and overlying vitreous with extension of fanlike projections towards the periphery. This lesion may blend imperceptibly with the surrounding RPE with an absence of RPE or choroidal atrophy. The lesion may be covered by a thickened gray-white retinal and preretinal tissue which may show contraction of the inner surface. There is generally absence of retinal detachment, hemorrhage, and vitreous inflammation.5 The Macula Society1 reported retinal vascular tortuosity within the lesion in 93% of patients, hyperpigmentation in 87%, slight elevation in 80%, epiretinal membrane formation in 78%, and exudation in 7%. Peripheral lesions14 appear as an elevated ridge concentric with the disc. Adjacent to the lesion, larger retinal vessels appear stretched, and there are few smaller vessels (Fig. 137.2). A dragged disc appearance has been described in patients with peripheral lesions.5,15 Combined hamartomas are almost always solitary, unilateral tumors; however, a few cases of bilateral involvement have been reported in association with neurofibromatosis.9,16–18
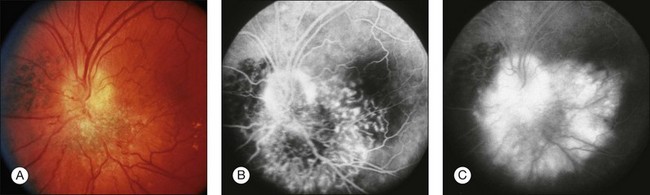
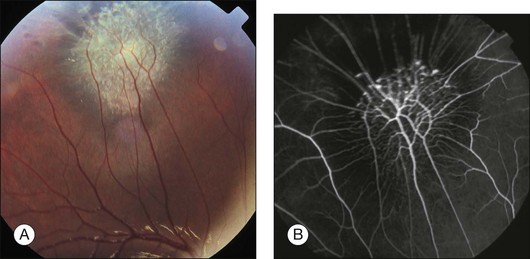
Associated ocular findings
Extensive disc or macular lesions may be associated with a relative afferent pupillary defect. Patients with macular involvement and decreased vision often have strabismus.7 Other findings include vitreous hemorrhage,10,11 preretinal neovascularization peripheral to the harmartoma,19 choroidal neovascularization at the margin of the lesion1,9,20 macular hole13 and peripheral hole formation.21 In addition, CHRRPE lesion have been associated with X-linked juvenile retinoschisis, optic nerve head pits, optic nerve colobomas and optic nerve head drusen.1,5,22
Systemic associations
Most patients with combined hamartomas do not have evidence of systemic disease; however, numerous reports describe an association with neurofibromatosis types 1 and 2.5,16–18,23–25 Bilateral CHRRPE lesions have been identified in patient with neurofibromatosis 1 and a majority of patients with bilateral combined hamartomas have signs of neurofibromatosis.9,16,18 In addition, CHRRPE lesions have been reported as a presenting sign in suspected neurofibromatosis. Of note, epiretinal membranes can also be present in patients with neurofibromatosis 2.25 Combined hamartomas have also been reported in patients with facial hemangiomas,5 incontinentia pigmenti,1 tuberous sclerosis,11 Gorlin Goltz syndrome,26 Poland anomaly,27 branchio-oculofacial syndrome,28 and juvenile nasopharyngeal angiofibroma.29
Diagnostic evaluation
Optical coherence tomography is an important modality for diagnosing and characterizing CHRRPE lesions. Early generation OCT images demonstrated an elevated lesion with high reflectivity of the inner retina, hyporeflective shadowing of the underlying tissue, and obscuration of the normal retinal layers.30,31 High-resolution spectral domain OCT imaging shows characteristic findings, including: peaked vitreoretinal traction, retinal disorganization, thickening of the retina at the level of the RPE and preretinal vitreoretinal interface abnormalities.32 OCT imaging can delineate the transition between the abnormal retinal interface and can guide surgical intervention. OCT is an important tool used to aid in the differential diagnosis and management of CHRRPE lesions. Ultrasonography is not useful in diagnosis of these minimally elevated lesions.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


