CHAPTER 170 Clinical Disorders of the Facial Nerve
This chapter describes clinical disorders of the facial nerve (Fig. 170-1). Most of the discussion is devoted to idiopathic paralysis (Bell’s palsy); however, disorders of the facial nerve associated with other diseases also are presented. The usefulness of electrodiagnostic testing and the medical and surgical management of Bell’s palsy are reviewed as well.
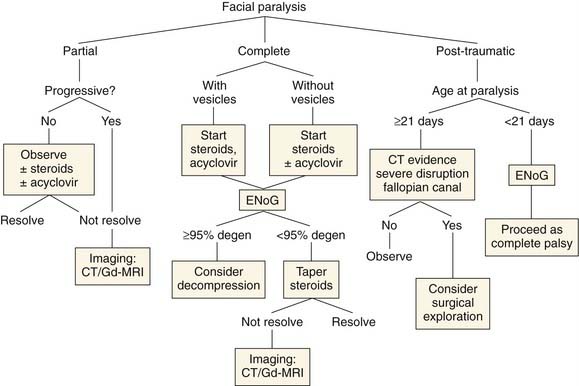
Bell’s Palsy: Spontaneous Idiopathic Facial Paralysis
Although older literature relegated Bell’s palsy to a diagnosis of exclusion, May and colleagues1 emphasized the diagnosis on the basis of specific clinical features. Taverner2 outlined the minimum diagnostic criteria for Bell’s palsy: (1) paralysis or paresis of all muscle groups of one side of the face; (2) sudden onset; (3) absence of signs of central nervous system (CNS) disease; and (4) absence of signs of ear or cerebellopontine angle disease. The differential diagnosis of facial palsy is shown in Table 170-1.
Table 170-1 Differential Diagnosis of Facial Paralysis
| Acute Paralysis | Chronic or Progressive Paralysis |
|---|---|
Modified from Adour KK, Hilsinger RL Jr, Callan EJ: Facial paralysis and Bell’s palsy: a protocol for differential diagnosis. Am J Otol. 1985;Nov(Suppl):68.
Incidence
The annual estimated incidence of Bell’s palsy is 20 to 30 patients per 100,000 population; it was 20.2 of 100,000 per year in the United Kingdom between 1992 and 1996.3 The incidence is greater in patients older than 65 years (59 of 100,000) and lower in children younger than age 13 (13 of 100,000).4–6 The male-to-female ratio for Bell’s palsy is approximately equal, except for a predominance in women younger than 20 years of age and a slight predominance in men older than 40 years.4 The left and right sides of the face are equally involved. Thirty percent of patients will have incomplete paralysis on presentation, and 70% will have a complete paralysis.7 Bilateral paralysis occurs in 0.3% of patients, and 9% have a history of previous paralysis.4 A family history of Bell’s palsy can be elicited in 8% of patients.4
Etiology
Proposed causes of Bell’s palsy include microcirculatory failure of the vasa nervorum,8,9 viral infection, ischemic neuropathy, and autoimmune reactions.10,11 Of these, the viral hypothesis has been the most widely accepted4,12; however, no virus has ever been consistently isolated from the serum of patients with Bell’s palsy.13 Thus, the evidence for the viral hypothesis is indirect, relying on clinical observations and changes in viral antibody titers. Furthermore, although the underlying cause may be viral, the immediate cause of the paralysis itself is debated to be viral neuropathy alone or ischemic neuropathy secondary to viral infection.14
Acute facial paralysis can occur as part of many viral illnesses, including mumps,15 rubella,16 herpes simplex,17 and Epstein-Barr virus.18,19 A viral cause for Bell’s palsy also is supported by the finding that it seems to be part of a polyneuritis syndrome in which the facial palsy is the most obvious cranial nerve involved. Careful neurologic examination will reveal other cranial nerve weaknesses in more than 50% of patients with Bell’s palsy.20,21 Adour22,23 found several affected cranial nerves in most patients (Table 170-2).
Table 170-2 Polyneuropathy in Bell’s Palsy
| Symptom | Incidence (%) |
|---|---|
| Hypesthesia or dysesthesia of the glossopharyngeal or trigeminal nerve | 80 |
| Hypoesthesia of C2 | 20 |
| Vagal motor weakness | 20 |
| Trigeminal motor weakness | 3 |
| Facial or retroauricular pain | 60 |
| Dysgeusia | 57 |
| Hyperacusis | 30 |
| Decreased tearing | 17 |
From Adour KK: Current concepts in neurology: diagnosis and management of facial paralysis. N Engl J Med. 1982;307:348.
A direct link between Bell’s palsy and viral infection has been difficult to establish. Serologic studies have examined both seroconversion and prevalence of antigens to suspected etiologic agents in patients with Bell’s palsy, especially HSV and VZV. The results have been conflicting and have been dependent on techniques used and populations studied.24,25 One reason for failure of these studies is that they sought for signs of an acute infection immediately preceding the onset of the Bell’s palsy. No seasonal predilection or epidemic clustering has been documented, however, so it is much more likely that Bell’s palsy is caused by reactivation of a latent virus rather than direct, communicable viral infection.
Morgan and colleagues26,27 and Vahlne and coworkers28 support the hypothesis that Bell’s palsy is a reactivation of VZV that may be initiated by several stresses, including heterotopic viruses (viruses other than herpesviruses) or physical or metabolic stress. Activation by other agents may explain the diversity and inconsistency of the antibody titers reported in various studies, as well as the epidemicity or clustering of cases. This reactivation can occur without a measurable antibody response to HSV.
The best evidence for reactivation of HSV as the mechanism for Bell’s palsy comes from several studies of the geniculate ganglion and facial nerve. Futura29 and Takasu30 and their coworkers have demonstrated HSV-1 DNA in both the trigeminal and geniculate ganglia of unselected autopsy specimens. Burgess and colleagues31 reported a single of case of a patient who died shortly after Bell’s palsy developed and in whom HSV-1 genomic DNA was identified in the geniculate ganglion of the facial nerve. Assay of archival control tissue for viral DNA yielded negative findings. In the most important study so far, Murakami and associates32 looked at endoneurial fluid and postauricular muscle from patients with Bell’s palsy or Ramsay Hunt syndrome and control subjects. Polymerase chain reaction (PCR) assay was used to test for HSV-1 DNA and VZV DNA, and the results were confirmed with Southern blot analysis. HSV DNA was detected in 11 of 14 patients with Bell’s palsy, whereas VZV DNA was not recovered from any. Conversely, VZV DNA was recovered from all the patients with Ramsay Hunt syndrome, whereas none had HSV-1 DNA. Unaffected control subjects had neither virus DNA. This study suggests a clean distinction between Bell’s palsy and Ramsay Hunt syndrome; however, other studies suggest more overlap.
Futura and coworkers33 studied 142 patients with both clinical Ramsay Hunt syndrome and acute facial palsy. Thirteen were diagnosed as having Ramsay Hunt syndrome on the first visit, and in another eight, vesicular lesions developed subsequent to their initial evaluation. Of the 121 with nonvesicular acute facial paralysis, 35 had VZV DNA detected in their saliva or serologic evidence of VZV reactivation. Yamakawa and colleagues34 demonstrated how technique-sensitive these studies are. Using two different PCR techniques, these investigators found high but differing rates of VZV DNA detection in patients with Ramsay Hunt symdrome.
A few animal studies also support a viral cause for Bell’s palsy. Ishii and coworkers35 induced facial paralysis in guinea pigs by inoculating the surface of the facial nerve with HSV-1. The severity of paralysis and subsequent degeneration were evaluated histologically and seemed to be worse if the epineurium was removed before inoculation. Of interest, these workers found greater extension of the virus centrally within the nerve than toward the periphery. Sugita and associates36 induced an acute transient facial paralysis in mice by injecting HSV into the auricles and tongues of mice. The paralysis started 6 to 9 days after inoculation and resolved after 3 to 7 days. Viral antigens were detected in the facial nerve, geniculate ganglion, and facial nerve nucleus. A spontaneously recovering ischemic facial palsy also can be induced in cats by embolization of internal and external maxillary arteries. The onset of the paralysis is immediate after embolization and resolves after 2 months.37
Although it is likely that the underlying disease in Bell’s palsy is viral polyneuropathy, an unresolved question is why such a profound effect on the facial nerve occurs, in contrast with the relatively minor and transient changes in the other cranial nerves. The major anatomic difference between the facial and other cranial nerves is in its long bony canal. Fisch38 measured the diameter of the fallopian canal throughout the temporal bone and found that the narrowest point was at the junction of the internal auditory canal and the labyrinthine portion, which he called the meatal foramen. The meatal foramen averaged 0.68 mm in diameter, whereas the remainder of the fallopian canal measured 1.02 to 1.53 mm. Fisch reasoned that edema in this narrow segment of the fallopian canal could cause damming of the axoplasmic flow within the facial nerve. This site of lesion in Bell’s palsy has been confirmed by clinical observation39,40 and by intraoperative neuronography.41,42 A similar site of lesion has been identified electrophysiologically in herpes zoster oticus.43
Histopathology
Fowler16 reported autopsy findings in a patient who died shortly after Bell’s palsy developed. The entire intratemporal nerve contained dilated and engorged veins and venules. Fresh hemorrhage was found within the internal auditory canal surrounding the facial nerve that extended as far as the geniculate ganglion. Fowler theorized that the ischemia resulted from microthrombi rather than vasospasm.
Reddy and associates44 examined a facial nerve 17 days after the onset of spontaneous paralysis. The nerve showed scattered degeneration of the myelin sheaths and axons. From 10% to 30% of facial nerve fibers were surrounded by phagocytic cells. The perivascular areas also showed inflammatory reactive changes and hemorrhage.
Proctor and coworkers45 examined a patient who died 13 days after the onset of facial paralysis. The facial nerve showed lymphocytic infiltration and phagocytosis of myelin by macrophages throughout the infratemporal course of the nerve. Initially, these findings were interpreted as evidence for a viral cause. However, when McKeever and colleagues46 reexamined the case, they emphasized that the inflammatory cells were most prominent where the facial nerve was surrounded by bone, especially in the labyrinthine portion of the fallopian canal, but not within the internal auditory canal. These investigators theorized that the histopathologic features suggested a compression-type injury with no evidence of vascular occlusion.
O’Donoghue and Michaels47 found degeneration of myelin sheaths and axon fibers throughout the intratemporal course of the facial nerve. The nerve was constricted at the meatal foramen, and an osteoclastic giant cell reaction caused resorption of bone around the geniculate ganglion. These workers interpreted the observed changes as consistent with a viral cause, but as pointed out by Jenkins and associates,40 these findings do not preclude edema and constriction of the facial nerve as the final event leading to the paralysis.
Podvinec48 found inflammatory infiltrates in the intratemporal portion of the facial nerve 6 months after the onset of the palsy, along with signs of wallerian degeneration and regeneration. He suggested that the persistence of infiltrates for this extended time may result from compromised circulation within the fallopian canal. Jackson and colleagues49 reported the histopathologic findings from the labyrinthine segment of the facial nerve in a patient 1 year after the onset of a total facial paralysis secondary to herpes zoster. A portion of the nerve within the auditory canal was essentially healthy; however, at the meatal foramen, the nerve became a mixed fibrotic and necrotic acellular mass.
Michaels50 described the histopathologic findings in an autopsy performed 11 days after the onset of Bell’s palsy. As in other studies, histopathologic features included demyelination, compression, bone resorption, and lymphocytic infiltration in the proximal labyrinthine segment of the facial nerve. Michaels cautioned against the overinterpretation of bulging of the nerve in the distal internal auditory canal, because it was found on both the healthy and the pathologic sides.
Another approach to the histologic evaluation of Bell’s palsy is with biopsy specimens obtained intraoperatively, including biopsy specimens of the chorda tympani and greater petrosal nerve. Chorda tympani specimens in acute Bell’s palsy have shown degeneration of myelinated fibers but no inflammatory response.51,52 Fisch and Felix14 examined greater petrosal nerve specimens obtained during middle cranial fossa facial nerve decompression. Their findings included degeneration and demyelination of large axons and lymphocytic infiltration. They found these results to be consistent with wallerian degeneration starting proximal to the geniculate ganglion, probably from the meatal foramen.
Central Nervous System Changes
Auditory symptoms, usually in the form of hyperacusis, may be present in up to 30% of patients with Bell’s palsy.53 The cause for these auditory symptoms is unknown. McCandless and Schumacher54 did not find evidence of a lesion affecting the cochlear nerve in patients with facial paralysis. Many researchers have attributed the hyperacusis to decreased damping of sound secondary to dysfunction of the stapedius muscle. However, most investigators have not found reduced contralateral stapedius reflex thresholds, suggesting that an absence of stapedial damping is not the cause.55
On the basis of these observations, many investigators have concluded that the auditory dysfunction results from CNS involvement.53 A few patients with Bell’s palsy have auditory brainstem response (ABR) abnormalities compared with ABRs in age-matched control subjects. These abnormalities include an increase in the wave I to V interval and the interaural difference for wave V. These changes usually are present bilaterally and resolve with recovery from the facial paralysis.55 These findings suggested a brainstem abnormality associated with Bell’s palsy that the study authors hypothesized is related to the reactivation of HSV. These findings should be interpreted with caution. Other investigators did not confirm the presence of such ABR abnormalities,56,57 and the findings have not been corroborated by somatosensory evoked potentials or visual evoked potentials.
Several investigators have looked for CNS changes in acute facial paralysis by studying the cerebrospinal fluid (CSF). Again the results are contradictory. Findings of elevated levels of myelin basic protein58 and pleocytosis59 in the CSF support CNS involvement in Bell’s palsy. Weber and coworkers60 found a normal total cell count, total protein concentration, blood-CSF permeability, and CSF–to–serum immunoglobulin ratios in most patients with Bell’s palsy. Only approximately 10% of these patients demonstrated some pathologic feature of the CSF, including a mild increase in blood-CSF permeability or pleocytosis. These workers concluded that their findings did not support the hypothesis that Bell’s palsy was a part of a cranial polyneuropathy.
CNS changes in Bell’s palsy also have been suggested by magnetic resonance imaging (MRI). Jonsson and associates61 found brain or brainstem changes in 5 of 19 patients with Bell’s palsy. These areas of increased signal intensity did not correlate with the facial nerve brainstem nucleus or the supranuclear pathways and were interpreted as indicative of unrelated vascular disease.
Electrophysiology and Testing
The multiple branches from the facial nerve, including the greater petrosal nerve, stapedial nerve, chorda tympani, and muscular branches, led to the development of topodiagnostic testing to localize the site of a facial nerve lesion. Schirmer’s test evaluates greater petrosal nerve function by measuring the lacrimal secretions accumulating on a piece of filter paper placed under the eyelid at the medial canthus.62 Stapedial nerve function can be evaluated with stapedial reflex testing. The chorda tympani nerve can be evaluated with taste testing or with the submandibular salivary flow test described by Magielski and Blatt.63 Unfortunately, the accuracy of localization with topodiagnostic testing has been disappointing.42 This is not a surprising finding in Bell’s palsy, in which the lesion is a diffuse demyelination throughout the nerve. However, topodiagnostic testing also has yielded disappointing results in investigation of tumors in which a sharply defined site of lesion was expected.64
Electrodiagnostic tests on the motor branches of the facial nerve also have been used to assess function and predict outcome. All of these tests have a similar shortcoming: Stimulation and recording are performed only distal to the lesion, rather than on opposite sides of the site of injury.65 Accordingly, such testing should be delayed because it will not demonstrate abnormalities until nerve degeneration has reached the site of stimulation, usually 4 to 5 days after injury.
Nerve Excitability Threshold
The use of electricity as a neurodiagnostic test was described by Duchenne in 1892 and was adapted to the facial nerve by Laumans and Jongkees.66 Application of the Hilger nerve stimulator is the method of facial nerve evaluation most commonly used by otolaryngologists. The extratemporal portion of the nerve is stimulated with a small-amplitude, pulsed direct current. The face is observed for the lowest current to produce a visible twitch. Although a threshold difference of more than 3.5 mA between the two sides has been considered to be suggestive of nerve degeneration, Gates67 found that with careful technique, low thresholds were obtained with percutaneous stimulation. The thresholds increased slowly with age and body weight, but nerve excitability thresholds greater than 1.25 mA in the upper division and 2 mA in the lower division were statistically abnormal.
Maximal Stimulation Test
A modification of the minimal excitability test, the maximal stimulation test, attempted to determine the difference between the strength and amount of contraction of the facial musculature caused by a supramaximal electrical stimulus.68,69 However, the maximal stimulation test is difficult to quantitate and is more subject to interobserver variation than is the minimal stimulation test.65
Electroneurography
Electroneurography (ENoG), also called electroneuronography, adds recording of the facial muscle action potential with surface or needle electrodes to the stimulation tests. Esslen70 introduced the use of bipolar surface electrodes for stimulation and recording of responses. The two electrodes are moved independently to produce the maximum amplitude of response. The response is evaluated by comparing the peak-to-peak amplitude of the maximum response obtained for the two sides of the face.
Electromyography
Electromyography (EMG) measures muscle action potentials generated by spontaneous and voluntary activity. It is distinct from other electrodiagnostic tests in which the activity is generated by active stimulation of the nerve. Denervation potentials are seen 10 days or longer after onset of the palsy; therefore, they are of limited value in determining early prognosis of facial paralysis. However, the loss of voluntary motor unit potentials within the first 3 to 4 days of paralysis suggests a poor prognosis.65 Conversely, retention of voluntary motor activity past the 7th day suggests that complete degeneration will not occur. Sittel and Stennert71 found that the detection of spontaneous fibrillation by needle EMG had an 80% accuracy in predicting a poor outcome 10 to 14 days after onset of paralysis. Unfortunately, this is too late to be useful in surgical decision making.
Nerve Conduction Velocity
Nerve conduction velocity can be measured between the stylomastoid foramen and the mandibular branch of the facial nerve. A strong correlation has been found between decrease in nerve conduction velocity and decrease in the compound nerve action potential measured by ENoG during the first 2 weeks after the onset of paralysis.72 Normal facial nerve conduction velocity ranges between 37 and 58 meters/second, and conduction velocities in this range were associated with uniformly good outcomes. Conduction velocities between 20 and 30 meters/second carry a 50% chance of significant residual paresis or synkinesis, and those less than 10 meters/second were associated with poor outcomes.
Interpretation of Electrical Tests
Excellent recovery of facial function is invariable when the decline in the compound action potential (CAP) as measured by ENoG does not reach 90%, and one half of patients in whom this level of degeneration is documented also have excellent outcomes.73,74 The problem is how to identify patients who will not achieve a good recovery, and at least a partial answer lies in the combination of ENoG with standard EMG. For patients who maintain recordable voluntary motor potentials despite a severe depression of the CAP, the prognosis is excellent.74 This finding may be explained by severe desynchronization of the motor units, causing a reduced or absent CAP.
Electromagnetic Stimulation of the Facial Nerve
Facial nerve testing with electromagnetic stimulation also has been explored. Benecke and colleagues75 were able to stimulate the nerve with cortical (supranuclear) electromagnetic discharges, demonstrating that the nerve could be stimulated trans-synaptically. Direct stimulation of the facial nerve with electromagnetic stimulation also has been described. On the basis of latencies of compound muscle action potentials from magnetic and electrical stimulation of the facial nerve, investigators concluded that the root entry zone of the facial nerve was the most likely site of excitation with magnetic stimulation.76 These data are tantalizing because the ideal test of the facial nerve would stimulate the nerve medial to the presumed site of the conduction block in the labyrinthine segment. Meyer and coworkers77 found an absence of electromagnetic responses to be the earliest sign of nerve conduction block in patients with Bell’s palsy, and that electromagnetic stimulation could differentiate between central and peripheral nerve lesions.
It is not yet clear, however, whether magnetic stimulation will be a useful predictive test, because significant discrepancies have been noted between the response to magnetic stimulation and clinical findings. Patients with incomplete palsies may exhibit complete loss of response to magnetic stimulation.78 Loss of response to electromagnetic stimulation occurs in more patients with Bell’s palsy than does loss of electrical stimulation, and this finding may persist after facial function has clinically recovered.75,79 Nowak and associates80 tested 65 patients with facial paralysis with both transcranial magnetic stimulation and electrical stimulation of the facial nerve. A severe reduction in muscle action potiental response was observed in both Bell’s palsy and herpes zoster and did not correlate with the clinical grade of the palsy. On the basis of current clinical information, electrical stimulation seems to add little to the diagnostic armamentarium in facial palsy.
Imaging of the Facial Nerve
Gadolinium-enhanced MRI has been used to study the peripheral facial nerve in Bell’s palsy. Enhancement of the facial nerve is a common finding in healthy persons, making evaluation of facial nerve findings on MRI difficult. The most consistent area of enhancement in Bell’s palsy and herpetic palsy is the distal meatal and labyrinthine segments.81,82 Sartoretti-Schefer and associates82 hypothesized that the gadolinium enhancement resulted from breakdown of the blood-brain barrier or from venous congestion in the epineurium. However, the location or degree of enhancement does not correlate with the recovery of facial movement.81,83 In a recent report that used high-resolution gadolinium-enhanced MRI with surface coils, Kress and colleagues84 were able to differentiate 3 patients with poor outcomes from 17 who recovered on the basis of the wide distribution and intensity of enhancement. The development of more detailed MRI techniques may prove useful in determining the prognosis of Bell’s palsy.85
Prognosis and Statistics
The prognosis for most patients with Bell’s palsy is excellent: 80% to 90% recover completely.6,53 Peitersen7 reported that none of 1505 patients followed without management had permanent complete paralysis, and that 17 had moderate to severe sequelae (contracture, synkinesis, palsy). Many large series of patients with Bell’s palsy have been analyzed to identify prognostic factors that have a significant impact on outcome. The most important such factor is whether the paralysis is incomplete or complete. The prognosis for affected persons in whom complete facial paralysis never develops is excellent: 95% to 100% recover with no identifiable sequelae.6,86
The many reports available in the literature are difficult to compare, because they use different criteria for inclusion and evaluation of outcome. Stankiewicz87 collated the outcomes from nine reports, including the large series of Park and Watkins88 and Peitersen.89 The overall results from these series were 53% complete recovery, 44% partial recovery, and only 3% no recovery. The results in the general population probably are better than those in these studies.
Other factors associated with poor outcome include hyperacusis; decreased tearing; age older than 60 years; diabetes mellitus; hypertension; and severe aural, anterior facial, or radicular pain.4,90 Abraham-Inpijn and associates,91 however, compared outcomes in 200 patients by use of several potential prognostic factors, including severity of the facial paralysis, mean arterial pressure, age, clinical or chemical diabetes mellitus, and history of hypertension. Only the severity of the paralysis at its maximum extent and the mean arterial pressure at presentation proved to be statistically significant. Peitersen7 noted a correlation between sequelae (synkinesis, contracture) and the interval between paralysis and the onset of recovery.
Management
The use of corticosteroids in Bell’s palsy was first proposed by Rothendler.92 Since then, corticosteroid therapy has become the most common approach to management of Bell’s palsy.93 Adour and Wingerd94,95 stated that although prednisone does not reduce the number of patients with eventual partial denervation or contracture or synkinesis, it reduces the number of patients with eventual complete denervation, therefore making the disease less severe. Numerous prospective and retrospective studies on the effectiveness of corticosteroids in Bell’s palsy have been reported. In a meta-analysis, Ramsey and colleagues96 found two studies with sufficient numbers of patients and rigor for analysis and concluded that treated patients had a 17% better chance of complete recovery than placebo-managed or untreated patients. Their analysis of a larger number of studies concluded that the odds of recovery with steroid treatment ranged between 49% and 97%, versus 23% and 64% for untreated patients. Consensus is lacking regarding the dose or duration of steroid treatment.
The case for steroid treatment in children in less clear. Neither Prescott97 nor Unüvar and coworkers98 found a beneficial effect of corticosteroids for Bell’s palsy in children. Salman and MacGregor99 came to the same conclusion in their systematic review of the literature.
Patients with Bell’s palsy commonly are treated with antiviral agents in addition to prednisone,100 although once again, definitive proof of efficacy is lacking. Although initial enthusiasm was high for the use of a combination of steroids and antivirals101,102 more recent studies have yielded negative results. Sullivan and colleagues103 conducted a four-armed, multi-institutional trial of prednisolone, acyclovir, a combined regimen of both agents, and placebo. Significant improvement in outcome was observed in both arms containing prednisolone, but no additional benefit from antiviral treatment was found. A similar outcome was described in a study reported from Japan.104 Current opinion does not support the use of antivirals in Bell’s palsy.
Surgical therapy is more controversial, because unlike with corticosteroids, it is associated with the possibility of significant additional injury. Early enthusiasm for transmastoid decompression of the tympanic and mastoid segments of the facial nerve has waned,105 and the procedure has been abandoned,1 because randomized trials showed no benefit1 and because of evidence that the site of the lesion is in the proximal labyrinthine portion of the facial nerve, which is inaccessible through the mastoid.38
The efficacy of surgical decompression of the meatal foramen and labyrinthine segment of the facial nerve in patients with poor prognosis as shown by ENoG testing is promising, but it has been difficult to assemble a large enough series to definitively establish its value in randomized trials. Fisch38 compared outcomes in 14 surgical patients with 90% degeneration or greater as shown by ENoG within 3 weeks of the onset of paralysis with those in 13 similar patients who refused surgery. He found a subtle but statistically significant improvement in the long-term facial recovery in the operative group. Sillman74 found a shift toward grade I and II recovery in patients with greater than 90% degeneration who underwent surgical decompression versus those managed with corticosteroids alone. In the most well-controlled study of middle fossa decompression of the facial nerve in Bell’s palsy, Gantz and colleagues106 evaluated the outcome when ENoG showed greater than 90% degeneration, and no voluntary motor unit potentials were found on electromyography performed within 2 weeks of onset of paralysis. Ninety-one percent of surgical patients achieved House-Brackmann grade I or II recovery, versus a 42% chance of these same results with steroids alone. Graham and Kartush107 described six patients with recurrent facial paralysis, one of whom had Melkersson-Rosenthal syndrome. None of these patients experienced recurrent facial paralysis after total seventh nerve decompression from the stylomastoid foramen to the internal auditory canal. Thus, surgical decompression remains in the armamentarium for the management of facial paralysis, but timing and appropriate patient identification remain difficult.
Persistent synkinesis can be a problem for many patients recovering from severe degeneration of the facial nerve regardless of the cause. Chua and associates108 performed a dose escalation study of botulinum toxin to determine the best treatment for reducing eyelid synkinesia and found the lowest dose, 40 units injected into the orbicularis oculi, to give the best reduction in synkinesia while avoiding ptosis. Borodic and coworkers109 reported similar results with botulinum toxin in a multi-institutional study.
Special Cases of Facial Paralysis
Ramsay Hunt Syndrome
Ramsay Hunt syndrome (herpes zoster facial paralysis) differs from Bell’s palsy, because it is associated with VZV infection (as shown by rising titers of antibodies to VZV) and the presence of skin vesicles on the pinna, retroauricular area, face, or mouth. Compared with Bell’s palsy, Ramsay Hunt syndrome generally causes more severe symptoms, and patients are at higher risk for the development of complete nerve degeneration.110
Varicella, or chickenpox, is the manifestation of primary infection by VZV (HSV, varicellae) in a nonimmune host. Herpes zoster is the manifestation of this same virus infection in a partially immune host. Serologic and epidemiologic data strongly suggest that varicella-zoster represents the reactivation of a latent virus rather than reinfection. After the primary infection, the virus probably travels to the dorsal root to extramedullary cranial nerve ganglia, where it remains dormant until it is reactivated. Reactivation generally occurs during a period of decreased cell-mediated immunity. VZV infection is the second most common cause of facial paralysis. The incidence of herpes zoster in patients with peripheral facial palsy is 4.5% to 9%.111 A Mayo Clinic study112 estimated the annual incidence of herpes zoster as 130 cases per 100,000. The incidence increased dramatically in patients older than age 60; 10% of this population had identifiable risk factors for decreased cell-mediated immunity, including carcinoma, trauma, radiotherapy, and chemotherapy. The increased incidence in the elderly is explained by an age-related decrease in cellular immune response to VZV.113
The severity of the paralysis is worse and the prognosis poorer in herpes zoster oticus than in Bell’s palsy. Peitersen114 reported full recovery in only 22% of patients, and Devriese115 found complete recovery in only 16%. As in Bell’s palsy, the recovery is predicted in part by the severity of the paralysis. Complete recovery occurred in only 10% of patients after complete loss of facial function and in approximately 66% after incomplete loss.111
The timing of the appearance of the vestibular eruption may have prognostic significance. In most cases, eruption and paralysis occur simultaneously. In approximately 25% of cases, the eruption precedes the paralysis; these patients have a higher likelihood of recovery.111 Patients with Ramsay Hunt syndrome also are more likely than patients with Bell’s palsy to have associated cranial nerve symptoms, including hyperacusis, hearing loss, and pain.116 Ramsay Hunt syndrome can be misdiagnosed on the basis of initial presentation. In approximately 10% of patients, the vesicular rash appears well after the initial facial paralysis, and many patients demonstrate a rise in antibody to VZV without ever exhibiting cutaneous or mucous membrane vesicles—so-called Ramsay Hunt sine herpete.
Severe ocular complications can occur with herpes zoster ophthalmicus. These complications include uveitis, keratoconjunctivitis, optic neuritis, and glaucoma and are almost always associated with involvement of the ophthalmic division of the trigeminal nerve. Herpes zoster ophthalmicus may be difficult to differentiate from the localized skin rash associated with HSV infection. Although both conditions may cause keratitis, differentiation between them is extremely important, because topical corticosteroids are used to manage herpes zoster but are contraindicated in HSV infection. Ophthalmologic consultation for biomicroscopy, staining, cytologic studies, and viral isolation studies may differentiate these two conditions.117 Adour94 stated that aside from concerns about ophthalmic involvement, the development of skin vesicles before or after initiation of prednisone does not contraindicate corticosteroid use.
Management of patients with herpes zoster, including cephalic zoster, consists of systemic corticosteroids.118 A specific benefit of corticosteroid therapy is a reduction in postherpetic neuralgia. The usefulness of corticosteroids in fostering recovery from the facial paralysis is controversial; however, early institution of corticosteroids seems to relieve acute pain, reduce vertigo, and decrease the incidence of postherpetic neuralgia.119,120
The antiviral agent acyclovir also is recommended to treat herpes zoster facial paralysis.121,122 Acyclovir is a nucleotide analog that interferes with herpes virus DNA polymerase and inhibits DNA replication. The drug is preferentially taken up by HSV-infected cells, making it nearly nontoxic to noninfected cells. Early return of facial function after acyclovir management has been reported by some investigators,121,122 but no beneficial effects were reported by others.123 At the least, treatment with acyclovir seems to lessen pain and promote resolution of the vesicles.



