CHAPTER 186 Cleft Lip and Palate
Classification of Cleft Lip and Palate
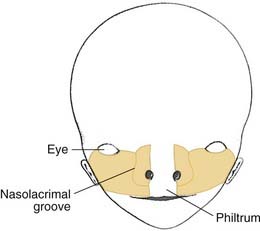
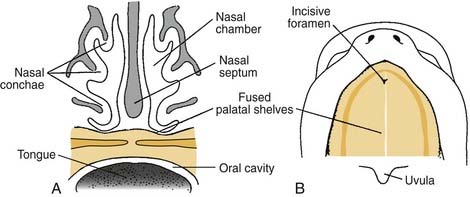
The incisive foramen divides the palate into the primary palate and the secondary palate. The secondary palate develops after completion of development of the primary palate, and extends from the incisive foramen anteriorly to the uvula posteriorly. The primary palate, which has the incisive foramen as its posterior border, consists of the premaxilla, lip, nasal tip, and columella.1
Cleft lip is classified as unilateral or bilateral, and its extent may be classified as complete or incomplete. A complete cleft involves the entire vertical thickness of the upper lip, and is often associated with an alveolar cleft, because the embryologic origins of the lip and primary palate are the same (Fig. 186-1). An incomplete cleft lip involves only a portion of the vertical height of the lip, with a variable segment of continuity across the cleft region. The variable continuous segment may present as a simple muscular diastasis with intact overlying skin or as a wide cleft with only a thin band of skin crossing the region of the cleft (Fig. 186-2). Simonart’s band is the bridge or bar of lip tissue, of variable size, that bridges the cleft gap. Simonart’s band usually consists of skin only, although some histologic studies have shown some muscle fibers to lie within the band.2 Unilateral clefts of the lip should include a designation of whether the right or the left side is involved.
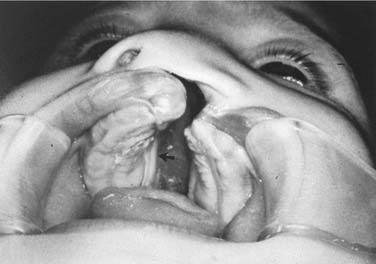
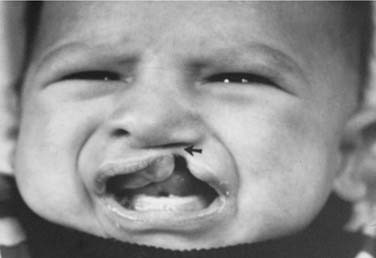
Figure 186-2. Arrow indicates Simonart’s band connecting two sides in a patient with incomplete cleft lip.
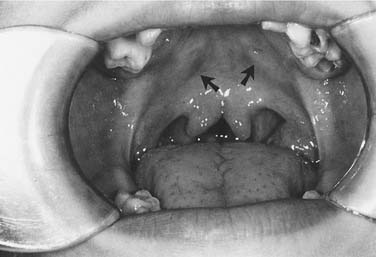
Palatal clefts also are described as being unilateral or bilateral, and their extent may be classified as complete or incomplete. In addition, cleft palates are classified according to their location relative to the incisive foramen. Clefts of the primary palate occur anterior to the incisive foramen, and clefts of the secondary palate involve the segment posterior to the incisive foramen. A unilateral cleft of the secondary palate is defined by a cleft in which the palatal process of the maxilla on one side is fused with the nasal septum (Fig. 186-3). A bilateral complete cleft of the secondary palate has no point of fusion between the maxilla and the nasal septum (Fig. 186-4). A complete cleft of the entire palate involves both the primary and secondary palate, including one or both sides of the premaxilla/alveolar arch, and frequently involves a cleft lip. An isolated cleft palate usually involves the secondary palate only, and has varying degrees of severity. The least severe incomplete cleft is the submucous cleft palate (SMCP) in which the underlying palatal musculature is deficient and inappropriately attached. Associated features include a bifid uvula, a zona pellucida (bluish midline region representing the muscle deficiency), and a notch in the posterior hard palate3 (Fig. 186-5). All three elements are not required for the diagnosis of SMCP, however.
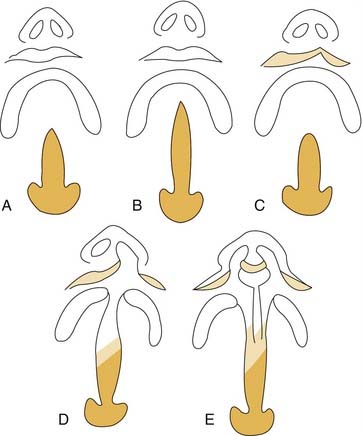
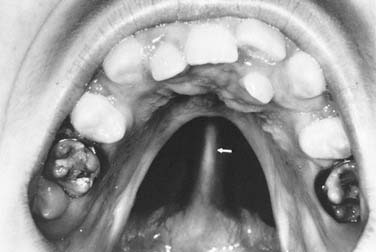
Figure 186-4. A patient with bilateral complete cleft palate. Arrow marks the nasal septum, which is not connected to either palatal shelf.
Embryology
For full appreciation of the variety of anatomic deformities that may be encountered in the patient with cleft lip and palate, an understanding of the normal embryologic development of the lip, palate, and nose is essential. At the end of the fourth embryonic week, the neural crest–derived facial prominences appear from the first pair of pharyngeal arches. The maxillary prominences are found laterally (Fig. 186-6). The frontonasal prominences, formed by the proliferation of mesenchyme ventral to the forebrain, form the upper border of the stomodeum. On either side of the frontonasal prominences are local thickenings of surface ectoderm which form the nasal placodes.1
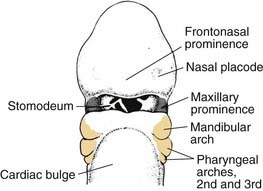
Figure 186-6. Lateral view of an embryo at the end of week 4, showing the position of the pharyngeal arches.
(From Sadler TW. Head and neck embryology. In: Sadler TW, ed. Langman’s Medical Embryology. 6th ed. Baltimore: Williams & Wilkins; 1990:315.)
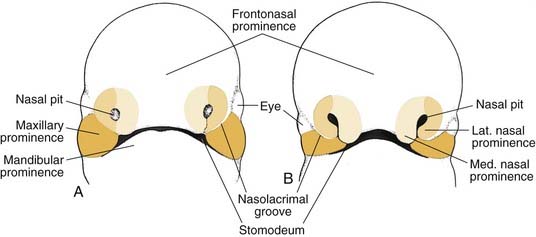
During the fifth week of embryonic development, the nasal placodes invaginate to form the nasal pits. This invagination process creates a ridge of tissue around the pit, called the lateral nasal prominences laterally, and the medial nasal prominences medially (Fig. 186-7).
Over the next 2 weeks, the paired maxillary prominences grow medially and approximate the paired medial nasal prominences (Fig. 186-8). Over time, fusion of the paired medial nasal prominences and paired maxillary prominences occurs, thereby forming the upper lip. The medial nasal prominences fuse to form the philtrum, medial upper lip, nasal tip, and columella. The maxillary prominences form the lateral aspects of the upper lip. The lateral nasal prominences are not involved in the formation of the upper lip, but form the nose (Fig. 186-9).1,3
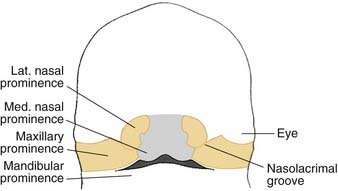
Figure 186-8. In a 7-week embryo, the maxillary prominences have fused with the medial nasal prominences.
(From Sadler TW. Head and neck embryology. In: Sadler TW, ed. Langman’s Medical Embryology. 6th ed. Baltimore: Williams & Wilkins; 1990:316.)
The nose is formed from five facial prominences: The frontonasal prominence forms the bridge, the fused medial nasal prominences form the tip and columella, and the lateral nasal prominences form the nasal alae (Table 186-1).
Table 186-1 Structures Contributing to the Formation of the Face
| Prominence | Structures Formed |
|---|---|
| Frontonasal* | Forehead; bridge of the nose, medial and lateral nasal prominences |
| Maxillary | Cheeks, lateral portion of the upper lip |
| Medial nasal | Philtrum of the upper lip, crest and tip of the nose |
| Lateral nasal | Alae of the nose |
| Mandibular | Lower lip |
* The frontonasal prominence represents a single unpaired structure, whereas the other prominences are paired.
From Sadler TW. Head and neck embryology. In: Sadler TW, ed. Langman’s Medical Embryology. 6th ed. Baltimore: Williams & Wilkins; 1990:315.
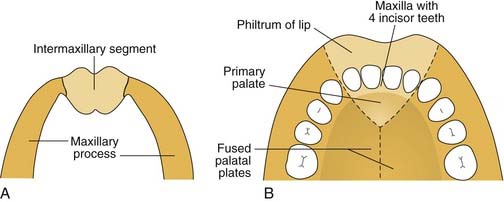
Palatogenesis begins at the end of the fifth week, with complete fusion occurring by 12 weeks of development. As the maxillary prominences grow and push the medial nasal prominences medially, the medial nasal prominences fuse not only at the surface but also at deeper levels (Fig. 186-10). Thus, the intermaxillary segment, or primary palate, which includes the central maxillary alveolar arch that houses the four incisor teeth and the hard palate anterior to the incisive foramen, is formed by the deeper levels of fusion of the two medial nasal prominences. Once the primary palate is completely developed, the secondary palate begins to develop.1,3
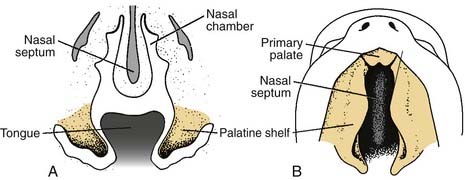
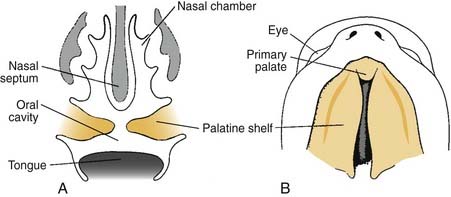
The secondary palate makes up the major portion of the palate. It is formed from the palatine shelves, which are two shelflike outgrowths of the maxillary prominences. In the sixth week of embryonic development, the palatine shelves are directed obliquely downward on either side of the tongue (Fig. 186-11). By the seventh week, the palatine shelves migrate inferomedially to lie horizontally above the tongue. It is in this horizontal position that the palatine shelves fuse in the midline to form the secondary palate (Fig. 186-12). The palatine shelves fuse with the previously formed primary palate anteriorly, and the nasal septum fuses with the newly formed secondary and primary palate. Palatal fusion occurs from anterior to posterior, beginning at the incisive foramen at 8 weeks of gestation and reaching completion by week 12 with uvular fusion (Fig. 186-13). The degree of clefting noted clinically is a consequence of the point in fetal development when the fusion process is interrupted.
Diagnosis
Prenatal Diagnosis
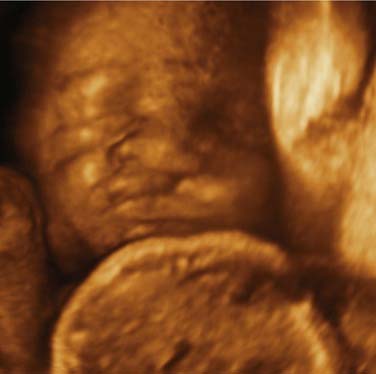
The advent of ultrasonography for use in prenatal care and improvements in imaging now permits antepartum diagnosis of cleft lip and palate4,5 (Fig. 186-14). Routine prenatal ultrasonography is now the standard practice in most communities. More reliable prenatal diagnosis can be made of cleft lip than of cleft palate.6 Diagnostic accuracy is improved if two-dimensional ultrasonography is combined with three-dimensional ultrasonography.7 The diagnosis can be made as early as 18 weeks, although the accuracy improves with the age of the fetus.8 It is estimated that 12% of these fetuses will have other anomalies.5 The parents frequently will want additional information and often are referred to cleft teams for counseling. Most families find antenatal counseling helpful in planning for care of the child with an orofacial cleft.9
Molecular Genetics of Orofacial Clefts
Clefts of the lip and/or palate are influenced by both genetic and environmental factors. Monozygotic twins demonstrate a concordance rate of 40% to 60% for clefting. Failure to reach 100% concordance in monozygotic twins suggests that genetics alone does not determine orofacial clefts. Embryology and genetics suggest that clefts of the primary palate, those that involve the lip with or without the remaining palate (CLP), arise from a different mechanism from that operative with clefts involving the secondary palate only (CP).10 Currently, approximately 70% of CLP patients are thought to be nonsyndromic, whereas 50% of CP patients are nonsyndromic.11 Syndromic clefts may be the result of a single gene transmission as occurs in autosomal dominant, autosomal recessive, or X-linked heredity. Facial clefting is a possible association in more than 200 syndromes.12 The most common syndrome associated with cleft lip is van der Woude syndrome. Lower lip pits and CLP are the characteristics features of this syndrome, although CLP and isolated CP can be seen in the same family and lip pits are not found in 10%. Genetic studies indicate that the gene responsible for this autosomal dominant disorder is on chromosome 1.13 A number of signaling molecules, transcription factors, and growth factors have been identified that are particularly important to the development of the lip and palate.14 Mutations to the gene, delay in expression of the gene product, or exposure to an environmental factor affecting the gene or its product may result in a cleft.
Several teratogens have been implicated in cleft development. These include alcohol,15 tobacco smoke,16 phenytoin,17 and retinoic acid.18 Meta-analysis did not find maternal age to be a factor.19 Multivitamin supplements lower risk for CLP and possibly isolated CP.20 Whether preconceptual folic acid supplementation alone can reduce clefting is controversial.21,22 Potential mechanisms of genetic, environmental, and biochemical interactions have been suggested and reviewed.20,23 Human teratogen studies should be interpreted with caution due to the potential for confounding variables and recall bias.
Genetic linkage studies have suggested a number of potential loci for clefts,24,25 though only loci on chromosome 6p have consistently demonstrated linkage to CLP.26,27 Linkage studies are limited by small numbers of families available for study. Transgenic mouse models and mouse strains with spontaneous clefts have identified candidate genes that may also affect human clefting. Mice deficient in the Msx1 gene, implicated in signaling between tissue layers during embryogenesis, all developed clefts of the secondary palate.28 Another putative mediator of orofacial clefting is transforming growth factor beta-3 (TGF-β3). Knockout of the TGF-β3 gene in mice results in clefts, but the addition of TGF-β3 to embryonic murine tissue culture stimulates palatal shelf adhesion.29 Studies of South American children with CLP suggest and interaction between Msx1 and TGF-β3 loci increasing cleft susceptibility.30 Transforming growth factor alpha also is considered to be a factor modulating CLP in humans.31
Epidemiology
Cleft lip and palate deformities are thought to be the most common congenital anomalies of the head and neck. Cleft lip with or without cleft palate must be distinguished from isolated cleft palate deformities because of different embryologic, etiologic, and epidemiologic factors.32 Cleft lip occurs in 1 in 1000 live births in the United States, whereas cleft palate deformity occurs in 1 in 2000 live births. Cleft lip deformities occur with the highest incidence among Native Americans (3.6 in 1000 births), Asians (2.1 in 1000), and whites (1 in 1000) and with the lowest incidence among blacks (0.41 in 1,000).12 By contrast, the incidence of cleft palate does not differ among ethnic groups, being reported as 0.5 in 1000 live births. Cleft lip occurs more commonly in boys than in girls, whereas cleft palate occurs more commonly in girls than in boys. Fusion of the palatine shelves 1 week later in girls than in boys is thought to be responsible for the higher frequency of cleft palate in girls.1
Recurrent risk rates for cleft lip with or without cleft palate and for isolated cleft palate have been determined and may be useful in counseling parents about the risk of cleft in future pregnancies.32 Two unaffected parents with one child affected with a cleft have a 4.4% chance of having another child with a cleft lip and palate and a 2.5% chance of having a child with isolated cleft palate. One parent affected with a cleft has a 3.2% chance of having a child with cleft lip and palate and a 6.8% chance of having a child with isolated cleft palate. Presence of a cleft in one parent and in one sibling is associated with a 15.8% chance that the next child will have a cleft lip or palate, and a 14.9% chance that the next child will have a cleft palate.32
Multidisciplinary Cleft Team
Comprehensive care of the child with cleft palate is best done within a team of health care professionals trained in the care of various disorders that accompany and develop as a result of the cleft lip or palate.33–35 This approach recognizes that no single discipline possesses all of the expertise needed for proper management of the many problems of these patients. The cleft team also will focus on the needs of the patient and family. Standards have been recommended by the American Cleft Palate–Craniofacial Association.36 Specialties that often make up a cleft care team are listed in Box 186-1. Members should meet face to face to discuss patient care at least six times a year. Continuing education for the care of patients with cleft deformities should be an integral part of membership in a cleft team.
Box 186-1 Specialty Members of a Cleft Care Team
Adapted from the American Cleft Palate–Craniofacial Association.
Team care begins when an infant is identified as having a cleft. The nursing staff or the pediatrician, within the context of a cleft care team approach, monitor feeding, growth, and development of the child. Parents are provided counseling, genetic information as needed, and verbal and written instructions regarding care plans. Most teams develop protocols for the management of children with cleft lip and palate deformities. These algorithms for care are based on the experience of the team members and assign care for anticipated needs of the child. A sample management protocol is outlined in Table 186-2. The otolaryngologist carries the roles of airway management, otologic care, and evaluation of velopharyngeal insufficiency and, in some teams, serves as the facial reconstructive surgeon.
Table 186-2 Example Protocol for Multidisciplinary Approach to Care of Patients with Cleft Lip and Palate
| Age Range | Intervention |
|---|---|
| Prenatal | |
| Neonatal (0-1 month) | |
| 1-4 months | |
| 5-15 months | |
| 16-24 months | |
| 2-5 years | |
| 6-11 years | |
| 12-21 years |
Nursing Care
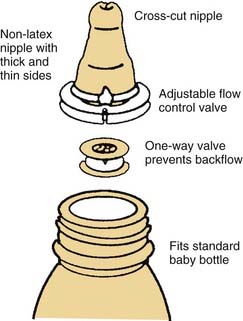
Infants with a cleft palate are limited in their ability to suck. The common cavity between the nose and the mouth permits air to leak as the infant tries to suck. Cleft lip alone usually does not cause feeding problems. Accordingly, a number of strategies have been devised and different feeders have been created for feeding the infant with a cleft palate. Generally, breast feeding is ineffective. Alternately, expressed (pumped) breast milk can be delivered using one of the specialized feeders. The three most commonly used cleft palate feeders are the Mead-Johnson, Haberman, and Pigeon bottles (Figs. 186-15 to 186-17). Each allows for parent-controlled delivery of the meal (expressed breast milk or formula). Parents and caregivers need to be taught how to properly use the feeder and observed for the first feed to insure proper use of the feeder.37,38 Trial and error may be needed to discover the best technique and feeder for the infant. Children with cleft palate generally swallow much more air during feeds. Frequent burping often is necessary; this should be explained to parents. Attention should be paid to the child’s growth. Following weight and length gains using standardized growth charts is very useful to ensure that the infant stays on the appropriate growth curves.

Anatomy
Anatomy of the Cleft Lip Deformity
The normal upper lip is divided into red and white components. The red lip is a mucous membrane, whereas the white lip is a cutaneous structure. The mucocutaneous junction at the vermilion border between the red and white lips is an important anatomic boundary that must be reconstructed meticulously in cleft lip repair for a natural-looking result. Failure to do so will draw attention to the irregularity at the vermilion border and create a poor cosmetic result.3,39,
In unaffected persons, the orbicularis oris muscle forms a complete sphincter around the oral cavity, providing the substrate for proper form and function of the lips and mouth. All patients with cleft lip deformities have muscular deficiencies and irregularities of varying degrees, leading to the abnormal appearance and function of the lip and mouth (Figs. 186-18 and 186-19). For proper correction of cleft lip deformities, it is essential not only to create symmetry of the lip superficially, at the skin level, but also to re-create the complete orbicularis muscular sling, for long-lasting cosmetic and functional results, and to re-establish complete mucosal coverage to ensure optimal healing and prevent distorting wound contracture. The muscle fibers in cleft lip deformities run in an inferior-to-superior direction along the margins of the cleft. They insert into the columella medially and along the nasal ala laterally. These fibers must be detached from their insertions and reoriented in a horizontal direction to bridge the cleft and create a complete muscular sling around the entire circumference of the oral cavity.3,39
Bilateral clefts also have abnormally oriented muscle fibers that run along the edges of the lateral aspect of the cleft (Fig. 186-20). Typically, the prolabial segment does not contain any useful muscle but is filled with connective tissue. In addition to the muscle and cutaneous irregularities, patients with bilateral cleft deformities have premaxillary and alveolar protrusion relative to the nasal septum. The premaxillary bony deformity may push the lip so far anteriorly and superiorly toward the nasal tip that the columella is severely diminished in strength and height and may even be obliterated completely (Fig. 186-21). There is often inadequate length to the medial crura; consequently, there is inadequate columellar skin. One of the major challenges in bilateral cleft repair is columellar reconstruction.3,39
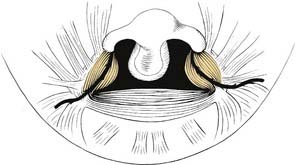
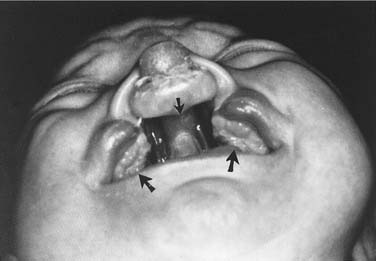
Figure 186-21. A patient with complete bilateral cleft lip and palate: Top arrow, protruding premaxilla; bottom arrows, collapse of alveolar arches.
The anatomic characteristics of the unilateral cleft lip nasal deformity include irregularities of the tip, columella, nostril, alar base, septum, and skeleton40 (Fig. 186-22). The nasal tip is deflected toward the noncleft side, with a relatively short medial crus and long lateral crus on the cleft side. In addition, the lateral crus of the lower lateral cartilage is caudally displaced on the cleft side. The columella is shorter than normal on the cleft side, and the columella lies on the non-cleft side due to the unopposed action of the intact orbicularis oris muscle. The nostril on the cleft side is horizontally oriented rather than the normal vertical orientation.17 Similarly, the nasal septum is deflected to the noncleft side. The alar base on the side of the cleft is displaced laterally, inferiorly, and posteriorly. Finally, there is a deficiency of maxillary bone on the cleft side, with the nasal floor often being absent.
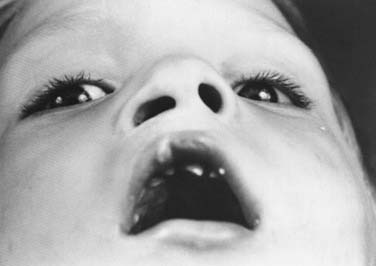
The bilateral cleft lip nasal deformity differs from the unilateral deformity in several aspects other than laterality. The degree of nasal deformity relates to the severity of the cleft lip deformity—whether the cleft lip is complete or incomplete. It also is affected by the degree of premaxillary protrusion. Bilateral cleft lip nasal deformities are symmetrical, making the repair of the nose somewhat simpler with regard to gaining tip symmetry. The challenging aspect of the bilateral cleft lip nasal deformity is the lack of adequate columellar tissue. This lack of tissue includes deficiencies in the length of the lower lateral cartilages, as well as deficiency of skin overlying these shortened cartilages. The tip usually is broad and flat, and the alae are laterally displaced, with resultant horizontally oriented nostrils.40
Cleft Palate Deformity
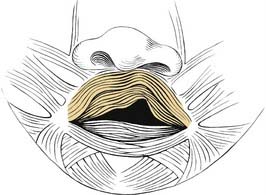
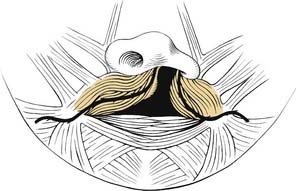
Five muscle pairs contribute to the soft palate. Normally, the levator veli palatini muscle has a transverse orientation, occupying the midportion of the soft palate, thereby creating a muscular sling for the velum.41 This muscular sling is the principal structural component in closure of the nasopharynx during speech and swallowing. Other muscles that contribute to the velopharyngeal sphincter include the palatopharyngeus, superior constrictor, and musculus uvulae. The musculus uvulae runs beneath the nasopharyngeal musculus and extends from the tensor aponeurosis to the base of the uvula.42 During speech, its contraction increases the midline bulk of the posterior edge of the soft palate. Deficiency of the musculus uvulae like that seen in submucous cleft palate may lead to velopharyngeal insufficiency and speech disorders (see Chapter 187, Velopharyngeal Dysfunction
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


