Chapter 152 Choroidal Osteoma
General considerations
The presence of bone within the eye is unusual and can develop in several clinical settings.1 Calcification can occur in tissues that are accessible to body fluids.2 The process of ossification occurs when an abundant blood supply is present to deliver osteoblasts to organize the architecture of Haversian systems.2,3 The choroid and the retinal pigment epithelium are the most common sites for bone formation within the eye. Intraocular ossification most often occurs as a dystrophic process in association with phthisis bulbi and in cyclitic membranes in the setting of chronic intraocular inflammation.2,4,5
The choroidal osteoma is an unusual form of benign intraocular ossification.1,6,7 In contrast to other types of intraocular ossification, it is found in otherwise healthy eyes. Interest in this tumor was stimulated by the observations of Dr Henry Van Dyk and associates who had the opportunity to study the clinical and histopathologic features of a case of an idiopathic osseous tumor of the choroid in 1975.8 The eye had been enucleated because of the clinical suspicion of an atypical malignant melanoma in a 26-year-old woman. In 1978, Gass and coworkers9 correlated the typical clinical manifestations and angiographic features of the choroidal osteoma in a series of four affected patients. Gass subsequently reported new observations in 15 patients with choroidal osteomas.10 Later, Shields and associates provided a comprehensive review on the subject of choroidal osteoma, including their own personal experiences with this tumor and Aylward and associates provided long-term follow-up of patients.1,11 Over nearly four decades, numerous clinical case descriptions of the choroidal osteoma have appeared in the literature.11–51
Definition and incidence
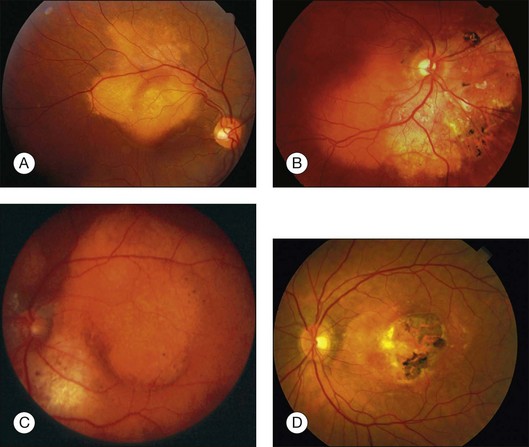
Choroidal osteoma is a benign tumor of the choroid composed of mature bone. It is typically found in healthy young females in the second or third decades of life (Fig. 152.1). However, some males,10,16,35,37,46,52 young children,21,23,24,34,38,40 and adults over age 30 years12,15,21,32,46,50 have been diagnosed with choroidal osteoma. There is no predilection for race. Most reported patients with this lesion are Caucasian, but several patients of African American24,31 and Oriental descent26,32,34,38,46,50,51 have been reported. The tumor generally occurs as a sporadic trait; however, a few cases of familial choroidal osteoma have been reported.21,23,40 The familial cases have been described in a mother and daughter,21 in a brother and sister,40 and in three siblings.23
Clinical features
Patients with a choroidal osteoma can be asymptomatic. Symptoms can include mild to severe visual blurring, metamorphopsia, and visual field defects corresponding to the location of the tumor.1,10 In one report, total blindness of both eyes was found in a young girl with bilateral choroidal osteomas.43
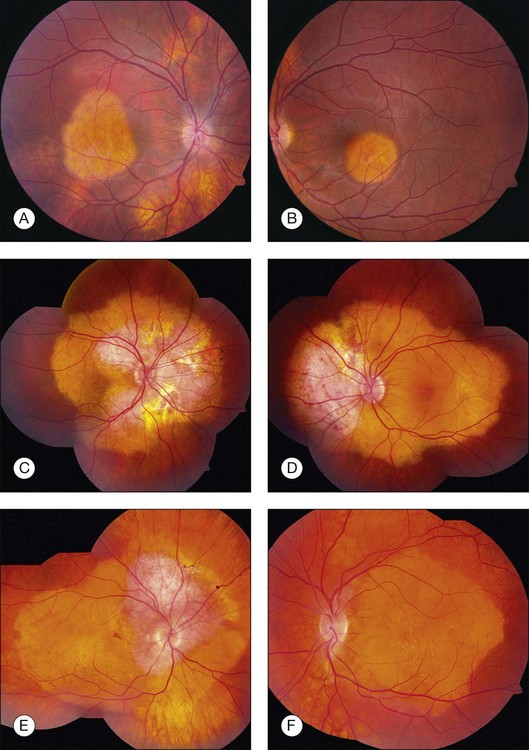
Choroidal osteoma is unilateral in approximately 75% of reported cases. In bilateral cases, the tumors can appear relatively symmetric or can be asymmetric, in different stages of development, growth, or decalcification (Fig. 152.2).10 Rarely, patients with initially unilateral choroidal osteoma, can later develop bilateral disease.30
The choroidal osteoma tends to be located in the juxtapapillary or peripapillary area, often with extension into the macula (Fig. 152.1). Rarely, this tumor is confined to the macular region only, without involvement of the juxtapapillary area.10,16,32,35,52 Choroidal osteoma is rarely seen in other locations of the eye and, to our knowledge, it has not been known to occur as a solitary lesion anterior to the retinal vascular arcades. Quite often choroidal osteoma is confused with idiopathic sclerochoroidal calcification, a benign, often multifocal, bilateral process that typically occurs anterior to the retinal vascular arcades, near the equator of the eye.53,54
The choroidal osteoma appears yellow-orange in color, particularly when the tumor is fully calcified. Over time, as decalcification occurs, the tumor becomes more pale yellow with overlying retinal pigment epithelial thinning and clumping. Choroidal osteoma ranges in size from approximately 2–22 mm in basal dimension and approximately 0.5–2.5 mm in elevation.1,10,11,20 In some cases, the bony tumor surface is irregularly excavated (Fig. 152.1) or elevated. The tumor shape is generally oval or round with characteristic well-defined scalloped or geographic margins. The margins can have blunt pseudopod-like projections. In some instances the tumor can be bilobed with two large plaques joined together by an isthmus.10,30
The vasculature within the choroidal osteoma is sometimes visible. Tufts of short branching vessels that originate deep within the tumor and emerge onto the tumor surface can be seen. They are especially prominent over the pale yellow portion of the tumor where the retinal pigment epithelium is thinned and depigmented.9 These large-caliber vessels are not neovascular tissue and are not associated with subretinal exudation or hemorrhage, or retinal pigment epithelial alterations.1,10 The overlying retinal vasculature and optic disc is characteristically unaffected by the osseous tumor and there are no associated vitreous or anterior segment abnormalities.
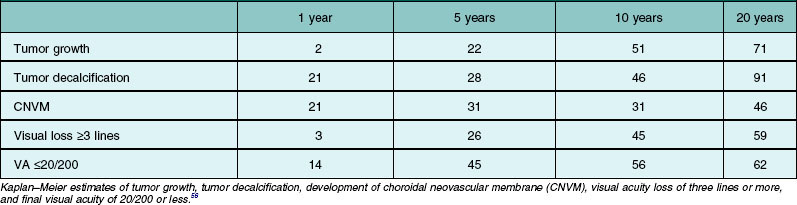
Many choroidal osteomas show evidence of slight enlargement in basal dimensions over several months to years (Fig. 152.3).11,38,41,44,48,55 Aylward and associates found tumor growth in 41% of 22 cases over a mean follow-up of 10 years.11 Shields and associates found tumor growth of 74 choroidal osteomas at 22% by 5 years and 51% by 10 years.56 In some cases, the tumor has doubled in size.10,41 Others have observed the development of a new choroidal osteoma in a previously unaffected eye.1,10
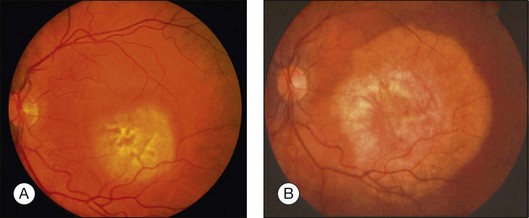
Trimble et al. reported a case of a choroidal osteoma in a 23-year-old woman, whose tumor enlarged slowly over 5 years.47 She developed a choroidal neovascular membrane that was treated with photocoagulation. Later, the tumor gradually decalcified over 18 months, leaving a bed of atrophy. Partial or complete decalcification of choroidal osteoma has been found to occur in 28% of cases by 5 years and 46% by 10 years.18,28,44,49,56 Subtle, gradual alterations in tumor color, surface pigmentation, and surface contour have also been observed.
Serous subretinal fluid can occur overlying choroidal osteoma.10 Subretinal fluid usually occurs over the macular portion of the osteoma. Careful ophthalmoscopy and fluorescein angiography should be performed for detection of choroidal neovascular membrane (CNVM) if symptoms or signs warrant.27
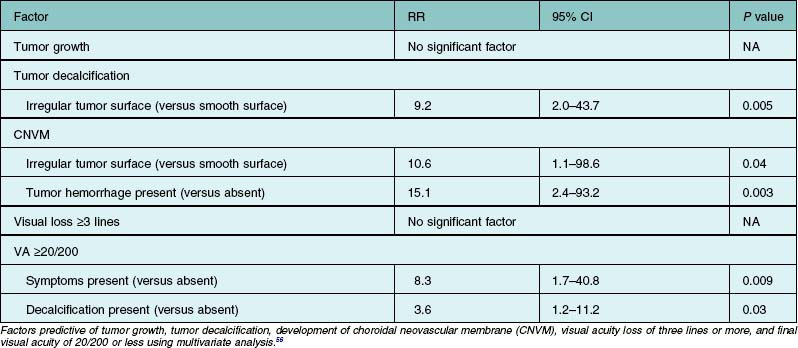
Choroidal neovascular membrane overlying osteoma has been found in 31% of cases by 5 years, 31–47% by 10 years, and 46–56% by 20 years (Fig. 152.4, Table 152.1).11,56 The CNVM is usually associated with subretinal fluid or hemorrhage, but it can be detected prior to vascular leakage as a slightly elevated gray-green subretinal tissue.15,37 Factors that predict the development of CNVM include irregular tumor surface and subretinal hemorrhage (Table 152.2).56 If the CNVM is extrafoveal then it can be treated with laser photocoagulation or photodynamic therapy.11,27 If the CNVM is in the fovea, the potential for subfoveal hemorrhage and disciform scarring often leads to a poor visual outcome.33,56 For subfoveal (and most extrafoveal) CNVM, we prefer antivascular endothelial growth factor (anti-VEGF) therapy monthly to resolve the membrane and then consolidation for extrafoveal CNVM with laser or photodynamic therapy.
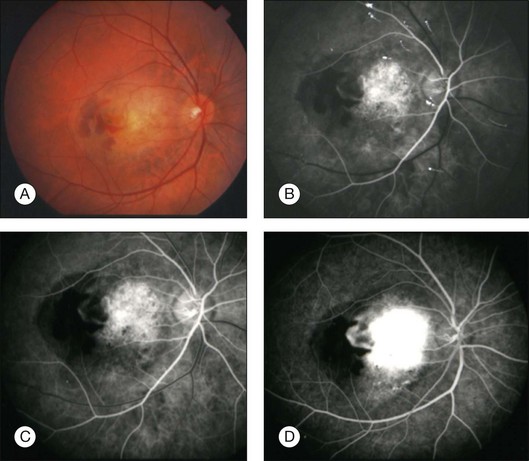
CNVM should be distinguished from the branching vascular tufts seen on the inner surface of the tumor. The latter are not associated with subretinal fluid, hemorrhage, or disciform scar and do not leak fluorescein on angiography as do the former. The reason for the occurrence of CNVM in eyes with choroidal osteoma is speculative. It is postulated that the thinned, degenerated retinal pigment epithelium – Bruch’s membrane complex overlying the osteoma develops breaks and neovascular fronds from the choroid grow beneath the sensory retina.27
Differential diagnosis
The ophthalmoscopic features of choroidal osteoma are classic. The most important lesions in the differential diagnosis include amelanotic choroidal melanoma, amelanotic choroidal nevus, metastatic carcinoma to the choroid, circumscribed choroidal hemangioma, disciform macular degeneration, posterior scleritis, idiopathic sclerochoroidal calcification, and choroidal cartilage.1,43,44,56–59
Amelanotic choroidal melanoma differs from choroidal osteoma in that it has a yellow-brown color, more elevation and a less well-defined margin. An amelanotic choroidal nevus can be relatively flat like a choroidal osteoma but a nevus differs in that it has less distinct margins and can have drusen on its surface. Choroidal metastasis tends to have indistinct margins and is often associated with a serous retinal detachment out of proportion to the size of the tumor.60 Furthermore, metastatic carcinoma usually occurs in middle age or older patients who have a prior history of malignancy (especially breast carcinoma). An irradiated choroidal metastasis may more closely resemble a choroidal osteoma because the irradiated metastasis appears flatter and atrophic and lacks appreciable subretinal fluid. Choroidal hemangioma can occasionally have overlying fibrous and osseous metaplasia that resembles osteoma.61,62 However, choroidal hemangioma is characteristically dome-shaped with smooth regular margins and overlying serous fluid with cystoid degeneration of the retina.
Macular degeneration is typically seen in older patients when it occurs in the spectrum of age-related macular degeneration or it is seen with other fundus findings of histoplasmosis when it occurs in the presumed ocular histoplasmosis syndrome. Macular degeneration is usually centered in the macular area and is less commonly in a juxtapapillary location. Posterior scleritis has a typical orange-brown color with indistinct margins.17 The symptom of pain and the findings of uveitis, choroidal folds, cloudy subretinal fluid, and ultrasonographic evidence of thickened sclera and choroid with retrobulbar edema aid in differentiating this entity from the choroidal osteoma.
Idiopathic sclerochoroidal calcification clinically appears as an irregular calcific plaque within the choroid (Fig. 152.5).54,59 The overlying retina and vitreous are normal. These findings resemble the clinical appearance of the choroidal osteoma. However, idiopathic sclerochoroidal calcification is not typically juxtapapillary and more often occurs outside the retinal vascular arcades, is usually multifocal, and can rarely occur in patients with other systemic diseases.53 The linear nevus sebaceous syndrome can manifest calcification within the choroid, with an appearance similar to choroidal osteoma, but histopathology has shown calcified scleral cartilage, not bone.58,63
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


