![]() 1
1 ![]()
Childhood Glaucoma
Kimberly S. Warren and Füsun Gökmen
Definition
How Is Childhood Glaucoma Defined?
Childhood glaucoma is a group of disorders characterized by improper development of the eye’s aqueous outflow system. Another term for this group of disorders is developmental glaucoma. Most developmental glaucomas are seen in childhood. Infantile glaucoma is any glaucoma occurring during the first several years of life, generally accepted as the first 3 years of life. Juvenile glaucoma is a nonspecific term referring to any type of glaucoma occurring in later childhood or the teenage years.
What Is Buphthalmos?
Buphthalmos means “ox eye” in Latin and refers to the marked enlargement of the globe that can result from any type of glaucoma present in infancy. Hydrophthalmia refers to the high fluid content of buphthalmic eyes.
How Is Childhood Glaucoma Classified?
We can divide childhood glaucoma into three major categories (Table 1–1): (1) primary congenital glaucoma, in which the developmental anomaly is restricted to a maldevelopment of the trabecular meshwork; (2) glaucoma associated with other ocular or systemic congenital anomalies; and (3) secondary glaucoma, which includes acquired ocular diseases that can cause glaucoma.1
| A. Primary congenital glaucoma (primary infantile glaucoma) |
| B. Glaucoma associated with congenital anomalies |
1. Late developing primary congenital glaucoma |
2. Familial hypoplasia of the iris with glaucoma |
3. Developmental glaucoma with anomalous superficial iris vessels |
4. Aniridia |
5. Sturge-Weber syndrome |
6. Neurofibromatosis |
7. Marfan syndrome |
8. Pierre Robin syndrome |
9. Homocystinuria |
10. Goniodysgenesis (iridocorneal mesodermal dysgenesis: Rieger’s anomaly and syndrome, Axenfeld’s anomaly, Peter’s anomaly) |
11. Loew’s syndrome |
12. Microcornea |
13. Microspherophakia |
14. Rubella |
15. Chromosomal abnormalities |
16. Broad thumb syndrome |
17. Persistent hyperplastic primary vitreous |
| C. Secondary glaucoma in infants |
1. Retrolental fibroplasia |
2. Tumors |
a. Retinoblastoma |
b. Juvenile xanthogranuloma |
3. Inflammation |
4. Trauma |
Is There Any Other Classification of Childhood Glaucoma?
Because some developmental glaucomas do not fit in any of the specific syndromes, an anatomic classification has been proposed (Table 1–2). Anatomic defects that are apparent on examination, form the basis of this classification. Maldevelopment of the anterior segment may involve trabecular meshwork alone or the trabecular meshwork in combination with the iris and/or the cornea. Identification of the type of anatomic defect helps in determining therapy and prognosis for the infant.2
When Is Microcornea Seen in Childhood Glaucoma?
Microcornea may be seen in a variety of congenital anomalies, including microphthalmos, nanophthalmos, Rieger’s anomaly, persistent hyperplastic primary vitreous (PHPV), congenital rubella syndrome, cornea plana, and sclera cornea. The corneal horizontal diameter is less than 10 mm. The eye is generally hyperopic but may be normal size, too. There is an increased risk of angle-closure glaucoma because of the shallow anterior chamber and narrow angles. Open-angle glaucoma can also be present.3
I. Isolated trabeculodysgenesis (malformation of trabecular meshwork in the absence of iris or corneal anomalies) |
A. Flat iris insertion |
1. Anterior insertion |
2. Posterior insertion |
3. Mixed insertion |
B. Concave (wraparound) iris insertion |
C. Unclassified |
II. Iridotrabeculodysgenesis (trabeculodysgenesis with iris anomalies) |
A. Anterior stromal defects |
1. Hypoplasia |
2. Hyperplasia |
B. Anomalous iris vessels |
1. Persistence of tunica vasculosa lentis |
2. Anomalous superficial vessels |
C. Structural anomalies |
1. Holes |
2. Colobomata |
3. Aniridia |
III. Corneotrabeculodysgenesis (usually has associated iris anomalies) |
A. Peripheral |
B. Midperipheral |
C. Central |
D. Corneal size |
What Conditions May Be Associated with Macrocornea in Childhood Glaucoma?
Macrocornea is seen in patients with Axenfeld’s syndrome and with X-linked recessive megalocornea. It is distinguished from the corneal stretching resulting from increased intraocular pressure (IOP) by the absence of tears in Descemet’s membrane.
Where Is the Pathology in Primary Congenital Glaucoma?
The iris and ciliary body have failed to recede posteriorly, and they overlap the posterior portion of the trabecular meshwork. This appearance is similar to an eye in the seventh or eighth month of gestation rather than at full term.4 An anterior insertion of the ciliary body muscle has also been found most specifically. The longitudinal and circular fibers of the ciliary muscle may insert into the scleral spur. The root of the iris may also insert directly into the trabecular meshwork. This malinsertion in the angle leads to blockage of aqueous humor outflow.5
The common defect is believed to arise from a developmental arrest during the third trimester of gestation of tissues derived from neural crest cells. The mechanism by which this developmental defect leads to aqueous outflow obstruction, in some cases, may be a paradoxical collapse of the trabecular meshwork and Schlemm’s canal in response to contraction of the ciliary musculatur, although other patients may have additional developmental abnormalities in the aqueous outflow system as the possible mechanism of glaucoma.6
What Is Barkan’s Membrane?
In 1955 Barkan7 described a persisting fetal membrane overlying the trabecular meshwork. Recent pathologic studies have not found evidence of Barkan’s membrane. This apparent membrane may be due to the observation of thickened, compact trabecular beams in the area of the meshwork.5
What Is the Cause of Elevated IOP in Congenital Glaucoma?
There is clinical evidence in childhood glaucoma that the obstruction to aqueous flow, with a resultant increase in IOP, is located at the level of trabecular sheets. Schlemm’s canal has been found to be open both histologically and clinically and does not appear to be the site of obstruction to aqueous flow.4,8,9
What Are the Congenital Anomalies Associated with Childhood Glaucomas?
Table 1–1 lists the congenital anomalies associated with childhood glaucoma. Each condition is described below.
What Is the Risk of Developing Glaucoma in Familial Hypoplasia of the Iris?
This condition is characterized by hypoplasia of the anterior iris stroma, a prominent pupillary sphincter, trabeculodysgenesis, and glaucoma. Glaucoma may occur any time from birth until late adulthood, but eventually will develop in almost 100% of cases. The hereditary pattern is autosomal dominant.10,11
What Are the Related Conditions with Anomalous Superficial Iris Vessels?
Irregular superficial iris vessels are generally seen with the distortion or absence of the superficial iris stroma and distortion of the pupil in newborn children with glaucoma. The cornea is usually hazy. These vessels should be differentiated from the normal radial iris vessels, which are straight and have no associated distortion of the iris tissue. Generally this is a bilateral disease.
How Often Is Aniridia Associated with Glaucoma?
Aniridia is a bilateral congenital anomaly characterized by marked hypoplasia of the iris, keratopathy, foveal hypoplasia, cataract, ectopia lentis, and optic nerve hypoplasia.12 Retardation of psychomotor development also may be evident. Trabeculodysgenesis of the anterior chamber angle or progressive pulling up of the residual iris stump and occlusion of the trabecular meshwork with synechiae formation are the proposed pathologies for the development of glaucoma. In most cases glaucoma does not develop until later childhood and sometimes does not develop at all.
Is Aniridia Hereditary?
Aniridia is most commonly a hereditary disorder transmitted as autosomal dominant. It also can occur sporadically, and approximately 20% of patients are found to have Wilms’ tumor. There is a specific syndrome caused by a partial deletion of the short arm of chromosome 11 and includes aniridia, Wilms’ tumor, mental retardation, and ambiguous genitalia.13
When Is Glaucoma Suspected in Sturge-Weber Syndrome?
Sturge-Weber syndrome is a flat facial hemangioma that follows the distribution of the fifth cranial nerve. A meningeal hemangioma (which can produce a seizure disorder), choroidal hemangiomas, and episcleral hemangiomas may also be present.14 The glaucoma is present when the facial hemangioma involves the lids or conjunctiva. Facial hemangioma is usually unilateral but may be bilateral. Glaucoma occurs usually in infancy but may not develop until early adulthood in some cases. A combined mechanism of isolated trabeculodysgenesis and elevated episcleral venous pressure presumably plays a role in the pathology.15
When Should Glaucoma Associated with Neurofibromatosis Be Suspected?
Neurofibromatosis (von Recklinghausen’s disease) is an autosomal-dominant disease characterized by multiple café-au-lait spots, neurofibromas of the skin and peripheral and central nervous system, and absence of a portion of the sphenoid bone or other skeletal defects.16 Ocular involvement includes nodules on the iris (Lisch nodules) and eyelids, ectropion uvea, optic nerve gliomas, retinal astrocytic hamartomas, and proptosis resulting from either optic nerve gliomas or herniation of brain tissue into the orbit.
Glaucoma associated with neurofibromatosis generally is seen with neuromas involving the upper eyelid or the eye itself. Isolated trabeculodysgenesis or synechial closure caused by neurofibromatous tissue can be the mechanism of pathology. A sheet of avascular dense tissue may arise from the periphery of the iris and extend anteriorly into the angle.
What Types of Glaucoma May Be Encountered in Patients with Marfan Syndrome?
This syndrome is characterized by arachnodactyly, congenital weakness of the aorta, aortic and mitral valve disease, scoliosis, hypotonia, and ocular abnormalities. It is usually autosomal dominant, but 15% of cases are sporadic. Ocular involvement consists of ectopia lentis, microphakia, myopia, megalocornea, hypoplasia of the iris stroma, retinal detachment and glaucoma.17 Two types of glaucoma are seen. Pupillary block glaucoma, secondary to malposition of the lens is one mechanism of glaucoma in these patients. The lens, usually is subluxated and held by zonules that are attenuated and often broken. Open-angle glaucoma can develop in later childhood. Iris processes can bridge the angle recess and insert well anterior to the scleral spur.18
What is Pierre Robin Syndrome?
This is a rare syndrome characterized by micrognathia, glossoptosis, cleft palate, and cardiac and ocular anomalies such as cataracts, high myopia, retinal detachments, microphthalmos, and childhood glaucoma.19,20
How Do Patients with Homocystinuria Develop Glaucoma?
Homocystinuria is an autosomal recessive disorder with a defect in the enzyme cystathionine synthetase. Patients present with light skin and hair color, osteoporosis, mental retardation, seizures, and ocular abnormalities such as retinal detachment and ectopia lentis. The lens usually is luxated inferiorly, but may move anteriorly and cause pupillary-block glaucoma.18
What Are the Different Presentations of Goniodysgenesis?
Axenfeld’s anomaly involves corneodysgenesis of the peripheral cornea and iris, whereas Rieger’s anomaly involves the midperipheral area. Central corneodysgenesis is present in Peter’s anomaly.
What Is Posterior Embryotoxon?
Posterior embryotoxon is a prominent anteriorly displaced Schwalbe’s line that may be present in an otherwise normal eye without glaucoma. On the other hand, extensive mesodermal strands in the angle may be accompanied by glaucoma. Axenfeld’s anomaly is usually bilateral, with an autosomal-dominant pattern of inheritance. About 50% of the patients may develop glaucoma in infancy or childhood.21
What Is the Difference Between Rieger’s Anomaly and Rieger’s Syndrome?
Rieger’s anomaly is a maldevelopment of iris and angle structures involving midperipheral iris adhesions to the cornea, hypoplasia of the anterior iris stroma, as well as pupillary abnormalities such as distortion of the pupil, polycoria, and correctopia. These abnormalities are usually present bilaterally, with an autosomal-dominant inheritance pattern. Other ocular abnormalities include strabismus, cataract, retinal detachment, macular degeneration, hypoplasia of the optic nerve, and chorioretinal coloboma.22
Rieger’s syndrome is the association of the above ocular abnormalities with systemic abnormalities. Dental and facial anomalies are most common and include hypodentia, microdentia, molar hypoplasia, and hypertelorism. Other systemic anomalies include short stature, heart defects, neurologic problems, empty sella syndrome, deafness and mental deficiency. Glaucoma occurs in approximately 50% of the affected individuals.23
What Are the Features of Peter’s Anomaly?
Peter’s anomaly is manifested as central corneal opacification with adhesions of the central iris to the posterior surface of the cornea. These iris attachments arise from the collarette and attach to the cornea where there is an absence of Descemet’s membrane and thinning of the posterior corneal stroma. In extreme cases the lens may adhere to the corneal endothelium and become cataractous.24 Another term for Peter’s anomaly is anterior chamber cleavage syndrome. Glaucoma occurs in about 50% of the involved eyes anywhere from infancy to later childhood.
What Is Loew’s Syndrome?
Loew’s syndrome is a sex-linked recessive disease characterized by aminoaciduria and mental retardation in male infants. Another term for this group of conditions is oculocerebrorenal syndrome. Ocular abnormalities most commonly are cataracts and glaucoma. Generally, isolated trabeculodysgenesis is observed microscopically.25
How Is Microspherophakia Suspected?
The finding of a small spheric lens whose edges are clearly seen through a mid-dilated pupil may be suspected as microspherophakia. High myopia with shallow chamber in a young person is a characteristic of this entity. Due to the laxity of the zonules, the lens may subluxate in the posterior chamber or move anteriorly, resulting in pupillary block glaucoma.26
What Are the Characteristics of Glaucoma Seen in Rubella Syndrome?
The glaucoma may be present in infancy and accompanied by features of isolated trabeculodysgenesis. Glaucoma can also result from iridocyclitis. If there is any evidence of inflammation, this mechanism must be suspected. Rubella keratitis causes deep and diffuse corneal clouding, which must not be confused with corneal edema resulting from glaucoma.27
What Are the Chromosomal Anomalies Related to Glaucoma?
Trisomy 21, trisomy 13–15, trisomy 17–18, Turner’s syndrome, and trisomy 2q may be associated with glaucoma.28,29
What Is the Mechanism of Glaucoma in Persistent Hyperplastic Primary Vitreous?
This condition results from failure of atrophy of the primary vitreous and its vascular structures, and typically occurs unilaterally in a microphthalmic eye. A retrolental fibrovascular membrane attached to the posterior lens and ciliary process draws the processes into the pupillary space. Progressive opacification and swelling of the lens may cause angle-closure glaucoma, whereas contraction of the membrane may push the lens forward.
What Are the Common Childhood Secondary Glaucomas?
The common childhood secondary glaucomas are enumerated in Table 1–1. Each condition is discussed below.
When Is Glaucoma Seen in Retinopathy of Prematurity (Retrolental Fibroplasia)?
Retinopathy of prematurity is associated with a history of prematurity of the newborn. It is bilateral and nearly symmetric. One theory suggests that oxygen therapy causes vasoconstriction in the peripheral retinal vessels initially. Ischemia may invite neovascularization, which may grow through the internal limiting membrane into the vitreous in advanced cases. Eventually, the development of these retrolental fibrotic membranes may cause a forward displacement of the lens and iris and cause angle-closure glaucoma with some degree of pupillary block.30
What Tumors May Cause Glaucoma in Children?
Retinoblastoma is the most common intraocular tumor of childhood. The tumor may invade the iris and trabecular meshwork area. Many patients develop rubeosis iridis and intractable neovascular glaucoma.31 Juvenile xanthogranuloma is an uncommon skin disease with its onset in infancy.32 Ocular involvement is seen as a vascular, yellowish white, solitary or diffuse mass of iris. Tumor involvement of the trabecular meshwork area and ciliary body may also occur. The most common cause for glaucoma is a spontaneous hemorrhage into the anterior chamber.
What Is the Most Common Inflammatory Condition Present with Glaucoma in Children?
Chronic iridocyclitis seen in juvenile rheumatoid arthritis is rarely associated with discomfort or redness. Many patients with this “white uveitis” may present with sequelae that may include glaucoma, cataracts, and band keratopathy.33 For other causes, see Chapter 8.
Is Traumatic Glaucoma Common in Children?
Children may present with traumatic glaucoma that is very similar to that in adults. For a complete discussion, see Chapter 13.
Epidemiology and Importance
How Common Is Childhood Glaucoma?
Childhood glaucoma, in all its forms, occurs in about 1 in 10,000 live births.34 Primary congenital glaucoma is not a common disease. It is estimated to affect less than 0.05% of ophthalmic patients.35 Disease is bilateral in 75% of cases. Male gender is found to have a higher incidence of the disease (65%). More than 80% of the cases are evident before the first year of life. It is the most common glaucoma of infancy, presenting as 1 in 30,000 live births.
Are There Any Genetic Considerations for Childhood Glaucoma?
The majority of cases of primary congenital glaucoma are sporadic. An autosomal recessive inheritance is reported in 10% of the cases.19,35 The penetrance rate varies from 25 to 100%. It is also reported that the penetrance of affected siblings may be between 3 and 11%.36 As boys were found to be affected more than girls, in this study inheritance pattern was believed to be polygenic. Genetic counseling is very helpful to the parents of affected children, as there is a 3 to 5% risk of another sibling being affected.37
What Are the Characteristics of the Genes Involved in Primary Congenital Glaucoma?
There are currently two genetic regions associated with the primary congenital glaucoma—GLC3A and GLC3B. The GLC3A region is on chromosome 2p21. Recently, mutations in the human cytochrome P4501B1 gene (CYP1B1) were identified in patients with GLC3A. No candidate genes have been reported for the GLC3B region, which is located on chromosome lp36. Both genes have autosomal-recessive transmission, and glaucoma is present before 3 years of age.38
What Are the Genes Associated with the Secondary Glaucoma?
The PAX-6 gene located on chromosome llp13 has been implicated in a number of ocular disorders.38
Diagnosis and Differential Diagnosis
How Is Childhood Glaucoma Diagnosed?
The examination and diagnosis of young children can be quite challenging. Childhood glaucoma has different presentations and can coexist with a variety of rare pediatric syndromes and congenital defects. The clinician must be aware of the different presentations of childhood glaucoma and have the knowledge to suspect the possibility of the disease in the absence of symptoms. Childhood glaucoma has been divided into three main classifications based on primary and secondary mechanisms: (1) primary, including primary infantile glaucoma and juvenile open-angle glaucoma; (2) glaucoma associated with congenital anomalies; and (3) secondary childhood glaucoma.39 Key elements to making an accurate diagnosis include obtaining a complete history, paying careful attention to the clinical presentation, and doing a thorough examination.
How Does Primary Infantile (Congenital) Glaucoma Present?
Primary infantile glaucoma typically presents with epiphora, photophobia, and blepharospasm. The presentation of congenital glaucoma is seen in children 3 years of age or younger. The parents will often report that the child is constantly tearing, is extremely light sensitive, and squints his or her eyes to avoid the light. These symptoms occur from corneal irritation secondary to epithelial edema caused by increased IOP.40 Buphthalmos, or “bull’s-eye”–like enlargement of the eye also can occur from the increased IOP and is recognized by an increased corneal diameter and progressive myopia. Buphthalmos occurs in children under 3 years of age because of elasticity and stretching of the ocular tissue. Additional findings include a cloudy cornea with breaks in Descemet’s membrane (Haab’s striae), anisometropic amblyopia, and strabismus.
How Does Juvenile Open-Angle Glaucoma Present?
In contrast to primary infantile glaucoma, juvenile open-angle glaucoma may have no external signs or symptoms. It presents after the age of 3 years and often goes undetected until advanced visual loss has occurred, with decreased central vision or extensive visual field loss. It can be detected early by screening exams if the IOP is measured and the optic nerve is evaluated.
What Is the Best Way to Examine a Child If Glaucoma Is Suspected?
Examination of the pediatric patient is often difficult and depends largely on the patient’s age and ability to cooperate. Children older than 4 years of age are usually able to cooperate with slit-lamp evaluation, tonometry, and optic nerve evaluation. Gonioscopy and visual field examination are more difficult to perform on young children, but some are able to cooperate by the age of 5 to 6. By the age of 8 to 10, most children are able to perform automated visual fields and a complete ophthalmologic examination. In the younger pediatric age group (4 years and less), a mild sedative such as chloral hydrate syrup (25 to 50 mg/kg body weight) can be used in the office to perform a slit-lamp examination, applanation, gonioscopy, and fundus examination. This is not always successful, and often general anesthesia is needed to properly evaluate the child. It is important to remember that most general anesthetics lower the IOP, so this measurement should be obtained as soon as possible once the child is asleep.41 Ketamine has been known to raise the IOP.42
What Are the Clinical Signs to Look for During the Examination?
It is important to know that not all childhood glaucomas have presenting symptoms, but one can make the diagnosis by clinical signs and evaluation. Increased IOP, corneal edema, increased corneal diameter, iris and corneal anomalies, gonioscopic anomalies, refractive errors, and optic disc cupping are key elements when making a diagnosis of childhood glaucoma. Normal IOP in children is slightly lower than in adults, but 20 mm Hg can be considered the upper range of normal. A pressure of 20 mm Hg or higher should alert the physician.43 The cornea should be examined by slit-lamp biomicroscopy for the evidence of corneal edema. Haab’s striae or breaks in Descemet’s membrane can be seen in congenital glaucoma as a result of increased IOP. The corneal diameter should be measured. This is best evaluated by using calipers and measuring the horizontal diameter. The normal cornea diameter in infants is 9.5 to 10.5 mm, reaching 12 mm by adulthood.44 The iris and cornea should be inspected for anomalies. This will help in the later classification of the type of childhood glaucoma. Developmental anomalies of the iris and cornea are not seen in primary infantile glaucoma, and if present should raise the suspicion of glaucoma associated with congenital anomalies. Gonioscopy can be evaluated by the Koeppe, Goldmann, or the four-mirror hand-held lens. The iridocorneal angle differs in childhood from that in adulthood. The angle is open but the trabecular meshwork is a smooth, homogeneous membrane extending from the peripheral iris to Schwalbe’s line. As the child ages the trabecular meshwork increases in pigmentation and becomes coarser.
The physician must know the difference between normal and abnormal angle features in children in order to correctly diagnosis congenital glaucoma. In congenital glaucoma the angle is open with an anterior insertion of the iris root into the trabecular meshwork. Glaucoma caused by trauma or inflammation may show angle recession or peripheral anterior synechiae, respectively. Refraction should be performed. A myopic shift may suggest glaucoma. The cup-to-disc ratio should be examined in all children evaluated for glaucoma. A ratio greater than 0.3 is unusual in normal infants but is often seen in infants with glaucoma.45 It is useful to know that if therapy is successful, the cup-to-disc ratio may decrease in size over time. This is not always true for adults.
The diagnosis of childhood glaucoma is not based solely on one finding being abnormal. It is a clinical diagnosis based on multiple signs and symptoms. If the physician is uncertain of the diagnosis, it is best to reexamine the child in a few weeks for confirmation.
How Does One Determine If the Child Has Primary Infantile Glaucoma or Another Form of Childhood Glaucoma?
The history and examination provide the answer. Primary infantile (congenital) glaucoma is diagnosed by increased IOP and by the finding of a high insertion of the iris on the trabecular meshwork without other ocular or systemic developmental anomalies. If other ocular or developmental systemic anomalies are present, the child is classified as having glaucoma associated with congenital anomalies. If the glaucoma is acquired due to a primary problem such as trauma, inflammation, or tumors, then it is classified as secondary.
Can Other Disease Processes in Children Mimic Childhood Glaucoma?
The most common clinical features for childhood glaucoma include excessive tearing, clouding of the cornea, large corneal diameter, elevated IOP, and increased cup-to-disc ratio. The first three are common in children less than 3 years of age, and the last two are found in all ages. There are many other pediatric disease processes that can present with similar symptoms. It is important for the ophthalmologist to be knowledgeable of these conditions so that the child can receive proper diagnosis and treatment.
What Are Other Causes of Excessive Tearing?
The most common cause of tearing in the infant is obstruction of the lacrimal drainage system. It can be unilateral or bilateral and may be accompanied by purulent discharge. Photophobia is not commonly associated with this problem. Another cause of tearing is conjunctivitis, either viral or bacterial, which is usually associated with purulent discharge and injection of the conjunctiva. Trauma with resultant corneal abrasions can cause excessive tearing in the infant and can easily be diagnosed by examination. A corneal dystrophy can cause ocular pain and tearing as seen in Meesman’s corneal dystrophy secondary to epithelial vesicles. Other causes may be recurrent epithelial erosions as seen in Reis-Buckler’s dystrophy.
What Are Other Causes of a Cloudy Cornea?
There are many causes of congenital cloudy cornea. Table 1–3 lists the causes of congenital clouding of the cornea. It is important for the physician to keep in mind the differential diagnosis for a cloudy cornea in the infant and not always to assume it is secondary to increased IOP.
I. Dystrophies |
1. Congenital hereditary stromal dystrophy |
2. Congenital hereditary endothelial dystrophy |
II. Dermoid |
III. Sclerocornea |
IV. Infection |
1. Rubella |
2. Herpes simplex virus |
3. Syphilis |
V. Metabolic (inborn errors rarely present at birth) |
1. Mucopolysaccharidoses |
2. Mucolipidoses |
VI. Trauma |
1. Forceps delivery with Descemet’s breaks |
VII. Anterior chamber cleavage syndromes—can have glaucoma associated |
1. Axenfeld-Rieger’s anomaly |
2. Peter’s anomaly |
3. Posterior keratoconus |
VIII. Congenital glaucoma |
IX. Congenital anterior staphyloma |
What Dystrophies Can Cause a Cloudy Cornea in the Infant?
Both stromal and endothelial dystrophies can cause a congenital cloudy cornea. Congenital hereditary stromal dystrophy presents at birth and is autosomal dominant. It has superficial corneal clouding with a flaky or feathery appearance of the anterior stroma.46 Congenital hereditary endothelial dystrophy may be autosomal recessive or autosomal dominant in nature. The autosomal-recessive form presents at birth with an edematous stroma, a thickened Descemet’s membrane, and nystagmus. The autosomal-dominant form is usually seen between ages 1 and 2 with photophobia and progressive corneal swelling.47
What Is the Difference Between a Dermoid and Sclerocornea?
A corneal dermoid is a choristoma that is composed of epidermis and epidermal appendages within a fibrous stroma.48 Most dermoids are found on the inferior temporal limbus and span approximately 8 to 10 mm. Dermoids extend into the corneal stroma and adjacent sclera. Limbal dermoids are seen in association with Goldenhar’s syndrome. Dermoids can cover the visual axis or produce a large degree of astigmatism. Either of these situations can cause amblyopia and vision loss. If either case is present, excision may be warranted. Sclerocornea is a congenital condition in which the cornea is opaque, resembling the sclera, and the limbus cannot be identified.49
What Infections Can Cause a Cloudy Cornea in Children?
Rubella is caused by a single-stranded ribonucleic acid virus. Congenital rubella is the result of a maternal infection during the first trimester of pregnancy. The classic triad of infants born with rubella include heart defects, deafness, and cataracts. A variety of ocular abnormalities can be seen, including pigmentary retinopathy, anterior chamber anomalies, glaucoma, and microphthalmos. The central cornea is often cloudy at birth, which is caused by either an absence of or ruptures in Descemet’s membrane or by congenital glaucoma.50 Congenital syphilis is also a well-known infectious cause of childhood cloudy corneas. Congenital syphilis is caused by the spirochete Treponema pallidum and is passed to the fetus in utero. The classic triad of congenital syphilis consists of interstitial keratitis, Hutchinson’s teeth, and deafness. The interstitial keratitis of congenital syphilis usually is a late manifestation, presenting after the age of 2 years. The interstitial keratitis is secondary to an inflammatory response to previously present spirochetes and is not due to an active infection. Both corneas are usually involved within weeks of each other. The child can present with similar symptoms of childhood glaucoma including tearing, photophobia, and blepharospasm. Perilimbal injection is also present, differing from congenital glaucoma. The inflammation is usually sectoral and involves the superior stroma. Keratic precipitates are typically present in the early stages. As the disease progresses, neovascularization develops in the deep stroma. As neovascularization becomes denser, the cornea appears pink and is referred to as a salmon patch. As the disease regresses, the cornea is often left scarred, and ghost vessels can be seen in the mid- to deep stroma.
What Metabolic Diseases of Childhood Can Cause Cloudy Corneas?
There are a number of inborn errors of metabolism that can cause congenital cloudy corneas. Fortunately, these disorders are rare, but they warrant discussing as a differential diagnosis in childhood glaucoma. These disorders are caused by an abnormal metabolism of carbohydrates and lipids. The major classifications include the mucopolysaccharidoses, the sphingolipidoses, and the mucolipidoses. Three mucopolysaccharidoses (MPSs) that cause diffuse clouding of the cornea include Hurler (MPS I-H), Scheie (MPS I-S), and Morquio (MPS IV) syndromes. Both Hurler and Scheie syndrome present with a cloudy cornea at birth and progress over the first 6 months, whereas Morquio syndrome does not show clouding of the cornea until after the age of 10. Fabry’s disease, gangliosidosis type I, gangliosidosis type II (Sandhoff’s disease), and Niemann-Pick disease are all disorders of sphingolipids. The most notable corneal changes seen in the sphingolipidoses consist of cornea verticillata, whorl-like lines in the corneal epithelium. This is a characteristic finding in Fabry’s disease. The mucolipidoses that present at birth with a cloudy cornea include MLS II and MLS IV. Cystinosis is an amino acid metabolic disorder that has an infantile, adolescent, and adult form. The infantile form is most severe, and cystine crystals can be present in the cornea at birth. This form of the disease is associated with renal failure in early childhood.51
What Are the Characteristics of a Cloudy Cornea Due to Birth Trauma?
Tears in Descemet’s membrane can be seen as a result of birth trauma due to forceps delivery. Classically these have been described as vertical tears, whereas Haab’s striae found in congenital glaucoma have a horizontal orientation. These are only generalizations, and either horizontal or vertical tears can be found due to each of the above causes. Not all tears secondary to trauma will cause diffuse corneal edema. If edema is present from trauma, it will usually resolve over the ensuing weeks spontaneously. The edema from childhood glaucoma will resolve only after the IOP has been successfully lowered.
What Are Other Causes of a Large Cornea?
Megalocornea and high myopia are two conditions other than congenital glaucoma and childhood glaucoma associated with ocular and congenital anomalies that can cause a large cornea in children. Megalocornea is an enlarged cornea that is nonprogressive. The cornea is clear and has normal histology. The diameter of megalocornea is 13 mm or greater in horizontal diameter. Ninety percent of patients are male, and the condition is usually bilateral. A sex-linked inheritance pattern is evident. Megalocornea is usually an isolated finding but can be seen in addition with cataracts, and glaucoma secondary to angle anomalies. Large corneas can also be seen in patients with high degrees of axial myopia. These children do not display the other signs of congenital or juvenile glaucoma.
What Are Other Causes of an Abnormal Optic Nerve?
It is important to recognize congenital causes of an abnormal optic nerve and be able to differentiate these causes from the enlarged cup-to-disc ratio found in the childhood glaucomas. It is often difficult to examine the posterior pole in the pediatric population. Every effort must be made to get a good look at the nerves. At times, evaluation under anesthesia is needed if an adequate exam cannot be obtained in the office. Congenital abnormalities include an optic nerve coloboma, optic nerve pits, hypoplasia, and tilting secondary to myopia. Optic nerve pallor and atrophy can also be seen in children, and the appropriate workup must be performed to ascertain an underlying cause. Physiologic cupping must be distinguished from glaucomatous cupping. This is difficult in the pediatric population, as most children do not perform well with the traditional visual field testing. Examination of family members can often be helpful to rule out physiologic cupping, and the new nerve fiber analyzers may become quite useful in the pediatric population.
What Steps Should Be Taken When Evaluating and Diagnosing a Child for Possible Childhood Glaucoma?
This section of the chapter has dealt with the initial presentation, signs and symptoms, physical examination, and differential diagnosis of childhood glaucoma. It can be a challenging diagnosis, and a flow chart has been developed to help aid the practitioner in making the correct diagnosis (Fig. 1–1).
Treatment and Management
How Is Congenital Glaucoma Managed?
The treatment for congenital glaucoma is surgical. Medical therapy alone is not sufficient in lowering the IOP in children. A study performed at the University of Ankara, Turkey, showed that medical therapy alone in congenital glaucoma reduced the IOP to less than 21 mm Hg in 11.8% in short-term follow-up and 9.7% in the long-term.52
The main surgical procedures of choice remain goniotomy, trabeculotomy, or trabeculectomy, with or without antimetabolites. Goniotomy was initially described by Barkan in the 1940s. This technique requires direct visualization of the trabecular meshwork with a surgical goniolens. A goniotomy knife is then inserted 1 mm anterior to the limbus through clear cornea. The blade is passed through the anterior chamber 180 degrees from the initial entry site. The blade is then used to incise one-third of the chamber angle. The incision is aimed at the abnormal layer of tissue in front of the trabecular meshwork. If the initial goniotomy procedure fails, repeat goniotomy may be needed. The trabeculotomy creates a direct communication between the anterior chamber and Schlemm’s canal. The technique was modified by Harms and Dannheim.53
The surgery involves creating a conjunctival flap similar to that performed in a filtering procedure. A partial-thickness limbal based scleral flap approximately 3 × 3 mm is then dissected. A radial incision is then made at the sclerolimbal junction until Schlemm’s canal is entered. This can be identified by a gush of aqueous humor or blood. A trabeculotome is then inserted into Schlemm’s canal and rotated so the arm tears through the trabecular meshwork and enters the anterior chamber. The trabeculotome is then inserted in the other direction and the same procedure is performed. The angle is opened 180 degrees with this technique. An additional scleral flap can be made 180 degrees away from the initial site to perform the procedure, for a total of 360 degrees. The trabeculectomy procedure performed is the same in children and adults. It involves conjunctival flap either limbal or fornix based, and a partial-thickness limbal based scleral flap of approximately 3 × 3 mm. A fistulizing technique is then performed. A peripheral iridectomy is made to prevent blockage of the fisula with iris incarceration. The scleral flap is then closed with 10-0 nylon sutures and the conjunctiva is reapproximated with a vascular needle. There has been recent discussion as to whether anti-metabolites should be use in the pediatric population in hopes of increasing the long-term patency of the filter. Other surgical procedures are performed in childhood glaucoma in situations where the above have failed or are not indicated. These procedures include tube-shunt surgery, with the use of valved and nonvalved implants; cyclocryoablation; cyclophotocoagulation; and ciliary-body endophotocoagulation.
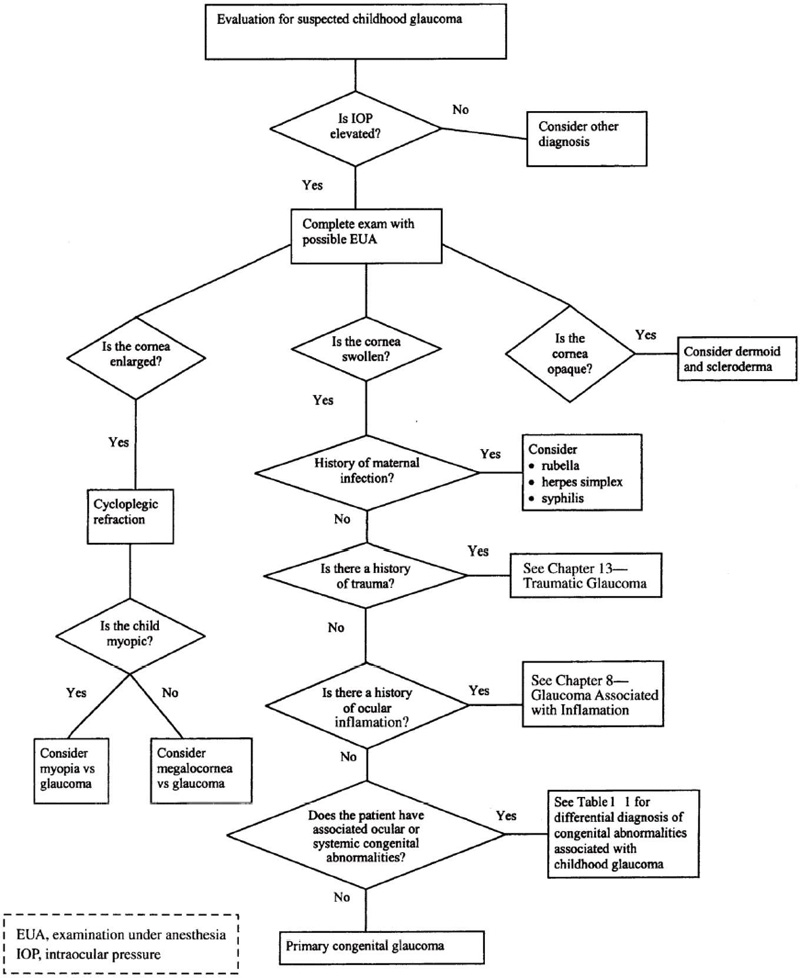
Figure 1–1. Algorithm for Evaluation of Childhood Glaucoma.
What Is the Best Surgical Treatment for Childhood Glaucomas?
The three principal initial surgical procedures for childhood glaucoma are goniotomy, trabeculotomy and trabeculectomy. Goniotomy and trabeculotomy have been the traditional choice for congenital glaucoma, with both having similarly high success rates. A study done at Moorfields Eye Hospital treating congenital glaucoma with initial goniotomy showed 93% of eyes controlled at 5 years.54 Goniotomy has both an advantage and disadvantage. The advantage is that it preserves conjunctiva if future surgery is needed, but the disadvantage is the need for a clear cornea to visualize the trabecular meshwork. The advantage to the trabeculotomy is that direct visualization of the trabecular meshwork is not needed, and if Schlemm’s canal cannot be identified, the procedure can be converted into a trabeculectomy. There has been recent interest in a combined trabeculotomy and trabeculectomy as the initial procedure in uncomplicated congenital glaucoma. A recent study performed in Saudi Arabia showed a 78% operative success in eyes with no coexistent anterior segment anomalies.55 Further studies comparing trabeculotomy, combined trabeculotomy and trabeculectomy, and trabeculectomy found that the results did not differ significantly.56 The best surgical choice for the initial treatment of congenital glaucoma is the technique that is most comfortable for the surgeon. Goniotomy is usually reserved for children of age 3 and under. Goniotomy and trabeculotomy are the first-line procedures in congenital glaucoma at a large referral center where the surgeons are experienced with these techniques. To the general ophthalmologist who is not experienced with the former procedures, the most appropriate primary surgery is the trabeculectomy.
What Is the Role of Mitomycin C in Filtration Procedures in Children?
At times a trabeculectomy is the procedure of choice in children. The trabeculectomy can be performed as the primary procedure or after previous failed trabeculotomy/goniotomy. In either case the question of the use of an antimetabolite must be considered. It is well known that trabeculectomy can fail in the pediatric population and is contributed to a thick Tenon’s layer and aggressive healing in children. A recent study evaluating the role of mitomycin C in childhood trabeculectomy showed a 1-year success rate of 76.9% in phakic patients.57 Interestingly, the same study revealed 0% success in aphakic eyes. Success was considered to be an IOP of less than 21 mm Hg with no need for antiglaucoma medication 1 year after the surgery. A similar study performed at Emory University School of Medicine showed a success rate of 67% ± 13% at 12 months and 59% ± 15% at 24 months. This same study identified aphakia and age of less than 1 year as significant risk factors for failure.58 The long-term complications such as bleb leaks with hypotony and endophthalmitis must be considered when using an antimetabolite in children.
What Is the Role of Tube-Shunt Procedures in the Pediatric Population?
Tube-shunt procedures are considered in the pediatric population in children with glaucoma refractory to medical and previous surgical intervention. A study of 18 patients performed at Wills Eye Hospital revealed a 72.2% success rate at 6 months.59 Success was considered to be an IOP between 6 and 21 mm Hg with or without glaucoma medication. At 2-year follow-up, the success rate was 44.4%, although five eyes (27.8%) had lost light perception and 12 of the 18 eyes underwent 28 additional surgical procedures to control IOP or manage tube-related complications. Tube-shunt procedures can be a surgical option for childhood glaucoma, but is usually reserved for refractory glaucoma and not as an initial therapy.
What Are the Follow-Up Considerations in Children with Childhood Glaucoma?
The child must be followed carefully to ensure that the glaucoma is stable and not progressing. Serial examinations are needed to ensure proper care. In congenital glaucoma the corneal edema is followed to ensure its resolution. A cloudy cornea can cause amblyopia and permanent vision loss. Once the corneal edema has resolved, the child is followed every 3 months to check the IOP, corneal diameter, and cup-to-disc ratio. Once the child is stable, examination can be performed every 6 months. It is important to keep in mind that amblyopia can result not only from an opaque cornea but also from anisometropia caused by a myopic shift. Careful attention must be paid to the refraction, and the child must be observed for any signs of a lazy eye. Many of these children require glaucoma medication, and the child must be watched to avoid systemic manifestation of the drugs. When using a beta-blocker, a complete history must be obtained, paying special attention to cardiac disease and a history of asthma. Pilocarpine can cause myopia and may be very difficult to tolerate, especially for children of school age.
Future Considerations
What Does the Future Hold for Early Detection of Glaucoma?
There have been recent discoveries concerning glaucoma and the field of genetics. Glaucoma loci have been grouped into three categories: primary open-angle glaucoma, primary closed-angle glaucoma, and congenital glaucoma. The congenital glaucoma group use the prefix GLC3. Primary open-angle glaucoma and primary closed-angle glaucoma use the prefix GLC1 and GLC2, respectively. There currently are two genes or genetic regions associated with congenital glaucoma. The congenital loci are GLC3A and GLC3B. The GLC3A region is on chromosome 2p21 and the gene is CYP1B.60 The GLC3B subtype has been located on chromosome lp36, but no gene has been identified yet.61 Secondary glaucomas have been linked to the PAX-6 and PITX2 genes. Aniridia and Peter’s anomaly have been linked to the PAX-6 gene located on chromosome 11p13.62,63 Axenfeld-Rieger syndrome type 1 and iridogoniodysgenesis type 2 have been linked to the PITX2 gene located on chromosome 4q25.64,65 Although still in its infancy, mapping and cloning of glaucoma genes hold a promising future. Early detection and treatment through genetic identification could halt advanced vision loss seen in childhood glaucoma.6
What Are the Future Challenges of Glaucoma Genetics?
The mapping and cloning of glaucoma-related genes have been progressing. It is anticipated that the actual number of glaucoma-causing genes will be much greater than the number currently known. There is considerable phenotypic variability that will make it difficult for clinicians and geneticists to associate clinical findings with the genes involved. However, as more genes are identified, the molecular pathogenesis of glaucoma may be better understood. Early genetic screening would allow the best treatment to be used for each patient. A greater number of patients can take advantage of early detection strategies to slow the progress of this insidious disease.6
Acknowledgment
This work was supported in part by a grant from Research to Prevent Blindness, New York, NY.
References
1. Schaffer RN, Weiss DI: Congenital and Pediatric Glaucomas. St. Louis: CV Mosby, 1970.
5. Maumenee AE: The pathogenesis of congenital glaucoma. Am J Ophthalmol 1959;47:827–836.
6. Shields MB: A common pathway for developmental glaucomas. Trans Ophthalmol Soc 1987;85:222–237.
12. Nelson L: Aniridia: a review. Surv Ophthalmol 1974;28:6:621–625.
14. Miller SJH: Ophthalmic aspects of the Sturge-Weber syndrome. Proc R Soc Med 1963;56:415–17.
15. Phelps CD: The pathogenesis of glaucoma in Sturge-Weber syndrome. Ophthalmology 1978;85:276–281.
21. Axenfeld T: Embryotoxon cornea posterius. Ber Deutsch Ophth Ges 1920;42:301–305.
22. Rieger H: Erbfragen in der Augenheilkunde. Graefes Arch Clin Exp Ophthalmol 1941;143:277–282.
29. Keith CG: The ocular manifestations of trisomy 13-15. Trans Ophthalmol Soc (UK) 1966;86:435–39.
36. Demenais F: Congenital glaucoma genetic models. Hum Genet 1979;46:305–313.
38. Freidman JS, Walter MA: Glaucoma genetics, present and future. Clin Genet 1999;55:71–79.
41. Duncalf D: Anesthesia and intraocular pressure. Bull NY Acad Med 1975;51:374–381.
45. New Orleans Academy of Ophthalmol: Symposium on glaucoma. St Louis: CV Mosby, 1981.
53. Harms H, Dannheim R: Trabeculotomy—results and problems. Bibl Ophthalmol 1970;81:121–131.
65. Walter MA, Mirzayans F, Mears AJ, et al. Autosomal dominant iridogoniodysgenesis and Axenfeld-Rieger syndrome are genetically distinct. Ophthalmology 1996;103:1907–1915.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


