Chapter 37 Childhood glaucoma
Classification
There are many classifications of childhood glaucomas. However, they can simply be classified as primary, where a developmental abnormality of the anterior chamber angle only exists, and secondary, where aqueous outflow is reduced due to congenital or acquired ocular diseases or systemic disorders (Box 37.1). Classification will change as we learn more about the genetic and biologic basis of each condition.
Clinical findings
A child suspected of having glaucoma will usually present in one of three ways:
1. Signs of elevated IOP (e.g. buphthalmos)
2. With a predisposing condition (e.g. aphakia), or
3. As part of screening where there is a family history of pediatric glaucoma.
Unique features of glaucoma in infancy
Generalized ocular enlargement
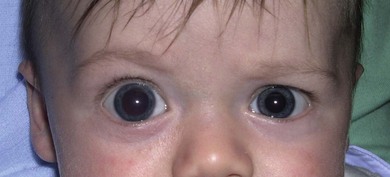
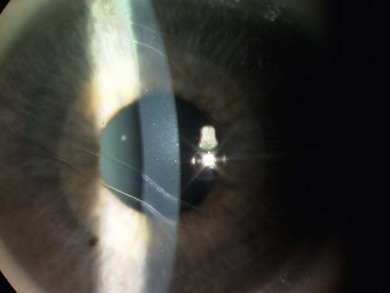
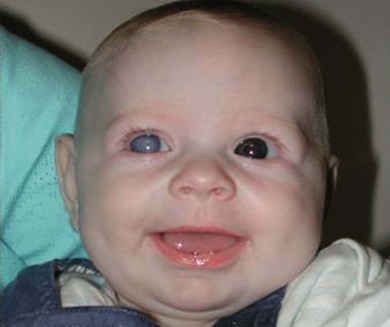
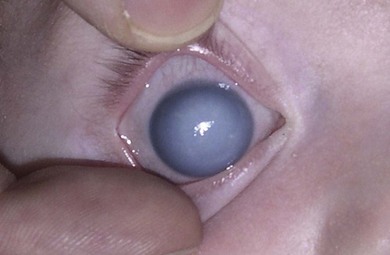
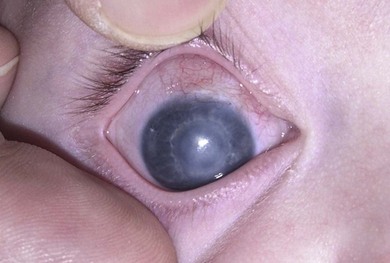
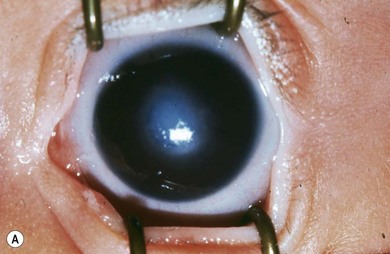
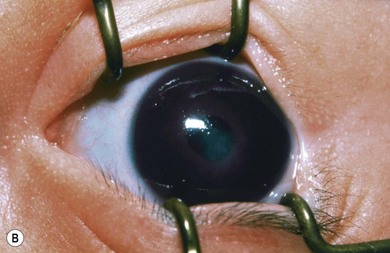
Buphthalmos refers to a prominent, enlarged eye due to elevated IOP from any cause in infancy (Fig. 37.1). The young eye is vulnerable to the effects of increased IOP due to corneal and scleral collagen immaturity. The potential for corneal enlargement usually ceases by the age of 3 although the sclera remains deformable until the age of 10. As the IOP rises, Descemet’s membrane eventually ruptures with one edge usually retracting as a scroll to form a ridge known as Haab’s striae (Fig. 37.2). As the underlying endothelium is torn, localized or diffuse corneal edema results from aqueous influx into the stroma causing a sudden cloudiness (Fig. 37.3). With successful lowering of IOP, corneal clouding clears first in the periphery (Figs 37.4 and 37.5). Photophobia may persist for several years with normalization of IOP due to the Haab’s striae.
Differential diagnosis
The differential diagnosis of glaucoma in children is broad. Remember that the IOP is normal and the signs will not be progressive in non-glaucoma cases. A list of differential diagnoses is outlined in Box 37.2.
Box 37.2
Differential diagnosis of childhood glaucoma
Corneal enlargement
Classification
Primary childhood glaucoma
Primary congenital glaucoma
Demographics
PCG is the commonest glaucoma in infancy but has an incidence of only about 1 in 10 000–20 000 live births in Western countries.1 The highest reported incidence is 1 : 1 250 in Slovakian gypsies. Parental consanguinity is responsible for the higher prevalence in certain ethnic and religious groups. PCG occurs more frequently in males than females at a ratio of between 2 : 1 and 2.5 : 1.
Genetics
GCL3A is the major locus for PCG, accounting for 85–90% of all familial cases. It has been mapped to the short arm of chromosome 2p21, the GLC3B locus to chromosome 1p36 and GLC3C to chromosome 14q24.3 with more loci speculated to exist.2 The primary molecular defect underlying the majority of cases of PCG is related to mutations of the CYP1B1 gene associated with the GLC3A locus3 but its frequency varies according to populations, ranging from 100% in Slovakian Roma to 20% in the Japanese. It encodes for enzyme cytochrome P4501B1 which is postulated to participate in the development and function of the eye. Genotype–phenotype correlations have been reported with specific mutations possibly associated with certain angle abnormalities.
Pathogenesis
The pathogenesis of PCG remains uncertain. The immature angle appearance results from the developmental arrest of tissues derived from cranial neural crest cells in the third trimester of gestation. Obstruction to outflow was thought to be due to the presence of an impermeable membrane (Barkan’s membrane); this has never been verified histopathologically. It is now thought to be due to thick, compacted trabecular sheets.4
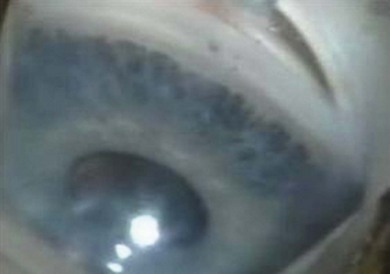
Gonioscopic findings
1. A thin and hypopigmented iris stroma
2. Peripheral scalloping of the posterior pigmented iris layer
3. Easily visible, hyperemic iris vessels with circumferential vessels running tortuously in the peripheral iris or on the ciliary body (Fig. 37.6).
Juvenile open angle glaucoma
Juvenile open angle glaucoma (JOAG) refers to patients who have no anterior segment abnormalities and usually present with glaucoma late in childhood but up to the age of 35. Often there is a strong family history of glaucoma. Up to 20% of patients have mutations of the myocilin/TIGR (trabecular meshwork inducible glucocorticoid response) gene at the GLC1A locus on chromosome 1q23.5 These patients typically have high pressures (40–50 mmHg) and respond well to prostaglandin analogs but often require an antimetabolite trabeculectomy.
Secondary childhood glaucoma
Anterior segment developmental anomalies (see Chapter 32)
Axenfeld’s anomaly [posterior embryotoxon (anteriorly displaced, prominent Schwalbe’s line) with attached iris strands] and
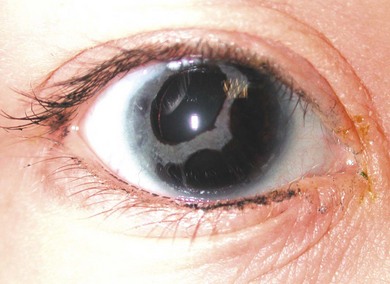
Rieger’s anomaly [these peripheral changes plus iris changes such as corectopia or iris thinning (Fig. 37.7)] and
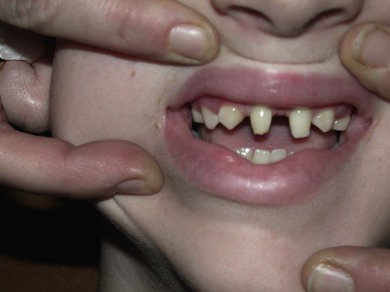
Axenfeld-Rieger syndrome refers to these ocular features in association with systemic anomalies such as hypertelorism, periumbilical skin folds, and dental abnormalities (Fig. 37.8). It is usually bilateral, asymmetric and inherited as an autosomal dominant trait. It shows genetic heterogeneity: several genes cause similar clinical phenotypes and, conversely, a single mutation may cause different phenotypes (variable expressivity). Several genes have been identified, the most common being RIEG1 at chromosome 4q25. It is also associated with FOXC1, RIEG2, and PAX6 gene mutations.
The risk of glaucoma with ASDA is 50%, so lifelong surveillance is indicated. Glaucoma usually occurs in childhood or young adulthood, very rarely in infancy and is due to an underlying trabeculodysgenesis. The IOP is often labile. The cause of outflow obstruction is due to the arrested maturation of angle structures. Medical therapy is indicated on diagnosis but surgery is required in the majority of cases, usually antimetabolite trabeculectomy. It is our impression that the more disorganized the anterior segment, the greater the risk of failure and the more potent the anti-scarring agent required. A potent antimetabolite is more likely to achieve IOP in the low teens and so significantly reduce corneal opacification (Fig. 37.9). Primary tube drainage surgery may be indicated in the most severe cases.
Chronic pupil dilation or an optical iridectomy (via a scleral approach to prevent further corneal scarring) should be considered rather than penetrating keratoplasty, which is associated with disappointing results.6 In Axenfeld-Rieger anomaly and iris hypoplasia where inheritance is often dominant, it is important to examine the patient’s parents and siblings.
Aniridia
Aniridia is characterized by bilateral variable absence of iris (see Chapter 32). Visual deficit in infancy is usually due to optic nerve and foveal hypoplasia. Later, loss of vision is due to glaucoma, cataract, ectopia lentis, and corneal surface abnormalities due to corneal epithelial stem cell dysfunction. Aniridia results from abnormal neuroectodermal development secondary to PAX6 gene mutations at chromosome 11p13. Inheritance is usually autosomal dominant although recessive transmission is possible; 30% of sporadic aniridia cases are associated with nephroblastoma (Wilm’s tumor), genitourinary abnormalities and mental retardation (WAGR) related to large deletions of 11p13, which encompasses PAX6 and the adjacent Wilm’s tumor locus. All sporadic cases must be screened regularly for renal abnormalities.
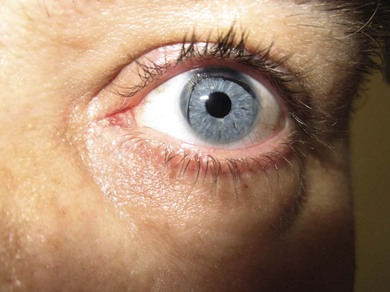
Glaucoma occurs in 50% of cases usually presenting in preadolescence or early adulthood. It may be due to progressive angle closure from the iris stump or be associated with an open angle. Medical therapy should always be first line treatment as this is safest but surgery is often inevitable. Goniotomy, both as therapy and prophylaxis, has been described but the latter is not widely practiced due to the potential risk of intraocular trauma. In the presence of a clear lens, we perform antimetabolite trabeculectomy with mitomycin C (MMC). If significant cataract is present or trabeculectomy fails, tube drainage surgery is indicated and can be associated with favorable results.7 Hypotony should be avoided due to the danger of lens-corneal touch, which will cause both cataract and corneal decompensation. Symptomatic relief of photophobia can be achieved with tinted spectacles, iris contact lenses, or a cosmetic iris implant inserted at the time of cataract surgery (Fig. 37.10).
Phakomatoses
The commonest phakomatosis associated with glaucoma is Sturge-Weber syndrome (encephalotrigeminal angiomatosis), a sporadic condition with a facial cutaneous angioma (port wine stain) present at birth, which affects the regions innervated by the first and second divisions of the trigeminal nerve (see Chapter 65). Choroidal hemangiomas occur in 40% of patients of whom 90% develop glaucoma. They are usually diffuse and can be easily missed on examination but identified by comparing the red reflex of both eyes or by ultrasound.
Other phakomatoses can be associated with glaucoma although with a lower incidence than Sturge-Weber syndrome (see Chapter 65):
1. Klippel-Trenaunay-Weber syndrome: triad of cutaneous hemangioma and varicosities involving one limb along with hypertrophy of bone and soft tissue. Most cases with glaucoma also have a facial nevus.
2. Neurofibromatosis: may present with iris abnormalities, e.g. ectropion uveae and glaucoma, before the systemic disease is apparent, especially if there is an ipsilateral lid plexiform neuroma.
Aphakic glaucoma
Its pathogenesis is uncertain. Both chemical (inflammatory cells, lens remnants, and vitreous derived factors) and mechanical theories (lack of ciliary body tension and trabecular meshwork collapse) have been proposed, combined with developmental reasons, i.e. arrest of postnatal angle maturation due to surgical insult. The incidence of glaucoma following childhood cataract surgery varies with duration of follow-up, ranging from 5% with simple aspiration8 to as high as 41% with lensectomy and vitrectomy with at least a 5-year follow-up.9 Risk factors for aphakic glaucoma such as microcornea, poor pupil dilation, early surgery, the need for secondary surgery and nuclear cataract are well documented. The role of posterior capsule integrity and intraocular lens implantation is still debated. Lifelong surveillance for glaucoma is crucial. If IOP measurement is difficult, monitor the optic disk appearance for progressive cupping frequently. Excessive loss of hyperopia or contact lens intolerance may be useful signs.
Aphakic glaucoma is refractory to treatment. Medical treatment is first line but only controls IOP in 50% of cases. Angle surgery does not provide good long-term control. Filtration surgery with MMC not only has a poor success rate10 but often excludes postoperative contact lens use in the presence of a thin, avascular cystic bleb due to the risk of endophthalmitis. Cyclodiode laser provides temporizing treatment with occasional long-term control after multiple treatments.11 However, it is difficult to titrate with marked inflammation and phthisis possible especially in microphthalmic eyes, in which case less energy should be considered. Furthermore, it may prejudice future surgery to failure or be associated with chronic hypotony. Drainage tube implants have the highest chance of long-term success and allow contact lens use.12 However, aphakic eyes have higher rates of complications, especially suprachoroidal hemorrhage, if hypotony occurs particularly if buphthalmic. Techniques to avoid catastrophic hypotony are mandatory.
Inflammatory glaucoma
This glaucoma is refractory to treatment, especially medical treatment. Angle surgery often requires medical treatment to control IOP.13 Antimetabolite trabeculectomy with MMC can be considered but recurrent ocular inflammation may compromise long-term success. Our preference is for primary tube drainage surgery especially if lensectomy is anticipated, or if aphakia is present. With any surgery, the risk of postoperative hypotony is high even in the absence of overdrainage due to potentially brittle aqueous production. Therefore, we prefer a smaller surface area drainage implant, e.g. Baerveldt implant (250 mm2) or Ahmed implant (184 mm2
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


