Chapter 24 Cell Death, Apoptosis, and Autophagy in Retinal Injury
Modes of cell death
According to the Nomenclature Committee on Cell Death 2009, “Cell death can be classified according to its morphological appearance (which may be apoptotic, necrotic, autophagic or associated with mitosis), enzymological criteria (with and without the involvement of nucleases or of distinct classes of proteases, such as caspases, calpains, cathepsins and transglutaminases), functional aspects (programmed or accidental, physiological or pathological) or immunological characteristics (immunogenic or non-immunogenic).”1 A cell is normally considered dead once it has passed an irreversible phase in the death process.
Apoptosis
Apoptosis, or programmed cell death, has received extensive study due to its critical role in development, tissue homeostasis, and pathology.2,3 Furthermore, apoptosis does not elicit an inflammatory response, thus allowing “physiological” cell death to take place without pathological consequences. Morphological features of apoptosis include rounding up of the cell, reduction in cellular and nuclear volume (pyknosis), nuclear fragmentation, modification of cytoplasmic organelles, plasma membrane blebbing, and engulfment by neighboring cells (Fig. 24.1).1 Apoptosis can be initiated by a variety of stimuli through two distinct pathways.4 The extrinsic pathway is triggered after the interaction of death receptors present on the cell surface with their cognate ligands (e.g., Fas ligand, tumor necrosis factor (TNF)-α and TNF-related apoptosis-inducing ligand (TRAIL)) and which can start the downstream executioner caspase (cysteine aspartic acid proteases) cascade within seconds of ligand binding. By contrast, the intrinsic pathway is initiated by a multitude of intracellular triggers, collectively called “stress signals,” which can include oxidative damage, deoxyribonucleic acid (DNA) damage, loss of cell–cell contact, growth factor withdrawal, hypoxia, and endoplasmic reticulum stress that all target the mitochondria and induce the release of proapoptotic factors to the cytosol that activate caspases. Excessive or deficient apoptosis is involved in numerous disease states. Readers requiring more detail are directed to the work of Green and Reed.2,3
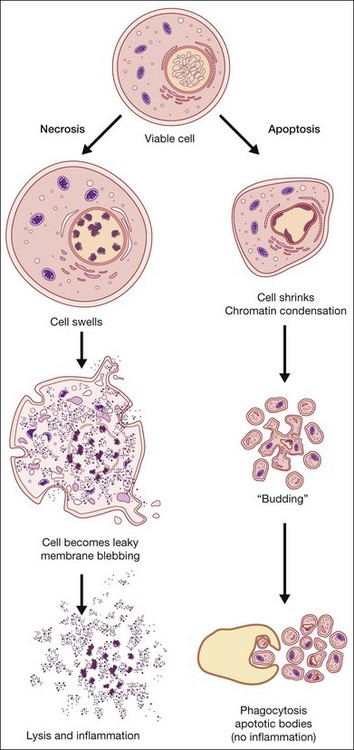
Necrosis
Necrosis has been defined as a type of uncontrolled cell death that can occur in response to infection, toxins, chemicals, injury, or lack of blood supply.2,5 Morphologically, necrosis is associated with cytoplasmic swelling (oncosis), rupture of the plasma membrane, swelling of cytoplasmic organelles, and moderate chromatin condensation (Fig. 24.1). The big pathophysiological difference between necrosis and apoptosis is inflammation. Necrosis culminates in the uncontrolled release of antigens which lead to activation of the immune system and inflammation whereas in apoptosis cell-bound bodies are formed which are phagocytosed by neighboring cells and there is an absence of inflammation. However, recent studies suggest that there is a molecular signaling network that can regulate the necrotic cell death pathway.6
Other
A number of other cell death pathways have been identified, of which autophagic cell death has gained some prominence. Increased autophagy (see below), such as that occurring under starvation, leads to self-destruction of intracellular organelles for provision of nutrients which, if starvation is not reversed, will culminate in self-destruction of cells and tissues. Autophagic cell death is morphologically defined as occurring in the absence of chromatin condensation, massive autophagic vacuolization, and little or no uptake by neighboring cells.1 Interestingly, autophagy is upregulated in a number of neurodegenerative diseases and thus may contribute to cell loss associated with these conditions. A number of atypical cell death modalities have also been identified and these are reviewed in Kroemer et al.1
Cross-talk between cell death pathways
Until recently, a requirement for gene expression was documented only for apoptotic and autophagic cell death. Interestingly, certain genes and their products, e.g., p53, Bcl-2 family proteins, and calpain, are important for both these modes of cell death.5–7 Recent work indicates that basal p53 activity suppresses autophagy, whereas the activation of p53 by some stimuli induces autophagy as well as apoptosis mediated by the Bcl-2 family proteins.6,7 Atg5 is essential for autophagy, but upon cleavage by calpains it has been reported that the truncated Atg5 associates with BclxL to promote cytochrome c release and caspase-dependent apoptosis.8 For many years, necrosis was regarded as the result of an accidental and uncontrolled process. However, accumulating evidence now suggests that necrotic cell death might also be mediated by a specific set of signal transduction pathways and degradative mechanisms. Similar to apoptosis, cell death with a necrotic appearance can contribute to embryonic development and adult tissue homeostasis. Some gene products, such as TNFR, CD95, TRAIL-R and RIP1, might trigger both apoptosis and necrosis, depending on interaction with other proteins.5 Moreover, there is cross-talk between these two cell death modalities. For example, inactivation of caspases might cause a shift from apoptosis to necrosis, or to a mixture of these two cell death modes.5 Thus cell death is not as easily defined as generally believed and there is considerable cross-talk between the different cell death mechanisms.
Autophagy and cell maintenance
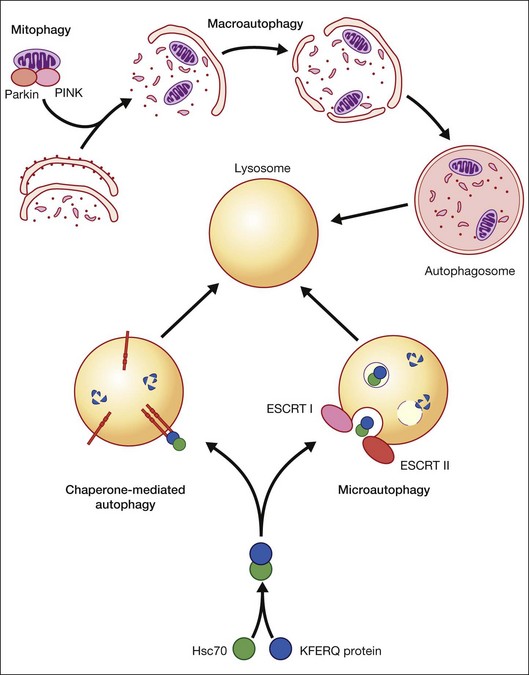
Autophagy is essential for cellular housekeeping and homeostasis through the sequestration and transfer of intracellular components (e.g., protein aggregates, organelles) to lysosomes for degradation.9–11 In mammalian cells, three primary types of autophagy have been reported: macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy (Fig. 24.2).12–16 These different types of autophagy are designed to nonselectively or selectively degrade different substrates and are regulated by different signaling pathways.
Macroautophagy is the best-characterized autophagy pathway that generally deals with larger substrates such as protein aggregates, intracellular pathogens, and dysfunctional organelles such as mitochondria (Fig. 24.2).9–11 The process of macroautophagy involves over 30 autophagy-related proteins (Atg) which regulate different stages of the autophagic response (Fig. 24.3). Macroautophagy is initiated by the sequestration of the cytosolic substrate into double membrane-bound phagophores which predominantly originate from the rough endoplasmic reticulum with possible contributions from the plasma membrane or mitochondria.17–20 The resulting autophagosome acquires endosomal and lysosomal proteins, ultimately maturing into a degradative autolysosome. The mTOR kinase complex is considered central to the signaling pathway of autophagy and can sense regulating conditions such as nutrient abundance, energy state, and growth factor levels.21,22 The PI3K-III complex consists of Vps34 and p150 and activators such as Beclin-1, Ambra1, ATG14, and UVRAG. This complex plays a crucial role in the induction of the autophagic process by generating PtdIns(3)P-rich membranes, which act as platforms for ATG protein recruitment and autophagosome nucleation.23 Antiapoptotic BH3 proteins such as BclxL and Bcl-2 bind to Beclin-1, having a negative impact on PI3K-III activity and autophagy. Elongation and completion of the phagophore are brought about by two ubiquitin-like conjugation systems, the Atg12-Atg5-Atg16 system and the Atg8-phosphatidylethanolamine (PE) system. Atg7 functions as an E1 enzyme in both systems, while Atg10 and Atg3 act as E2 enzymes for Atg12 and Atg8, respectively.24 The C-terminus of Atg8 is cleaved by Atg4 which primes the protein for conjugation to PE. The Atg12–Atg5–Atg16 complex recruits Atg8-PE to the elongating phagophore.25,26 At least eight different Atg8 orthologs belonging to two subfamilies (LC3 and GATE-16/GABARAP) occur in mammalian cells. LC3s are involved in elongation of the phagophore membrane whereas the GABARAP/GATE-16 subfamily is essential for a later stage in autophagosome maturation.27 The N-termini of LC3 and GATE-16 are required for autophagosome–lysosome fusion.28 Once in the lysosomes, substrates are degraded by the repertoire of lysosomal enzymes.29 It has been proposed that the autophagic elimination of mitochondria has its own specialized pathway.16 Critical to this process are the proteins PINK1, the E3 ubiquitin ligase, Parkin and possibly p62, a protein that binds to ubiquitin and LC3. PINK1 binds to uncoupled mitochondria which then facilitates the recruitment of Parkin which leads to ubiquitination of mitochondrial surface proteins. The ubiquitinated mitochondrion is then sequestered into the autophagosome through the likely actions of p62 and LC3.
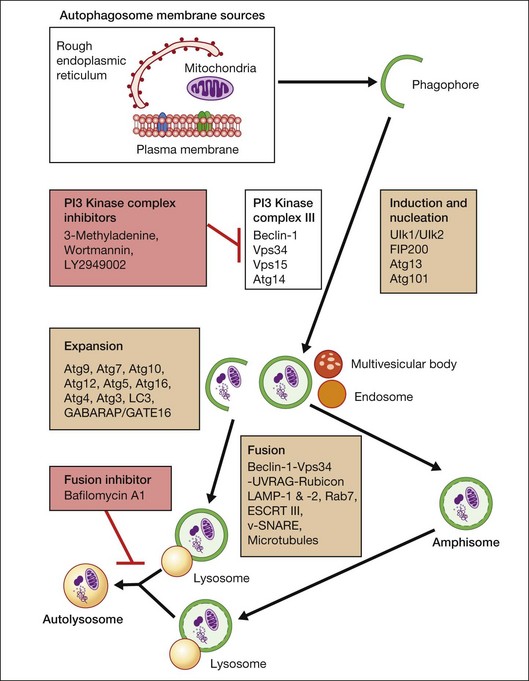
Autophagy can be activated by nutrient deprivation and environmental stress. For example, amino acid starvation and reactive oxygen species can stimulate autophagy.30,31 Recently, a distinction has been made between starvation- and stress-induced macroautophagy, also referred to as quality control autophagy. It has been observed that autophagic deficient cells tend to accumulate p62-rich aggregates, which in turn cause Nrf2 to be activated after separation from its interacting partner Keap1, which allows Nrf2 to mount an antioxidant response.32 In addition, histone deacetylase (HDAC)6 stands out as a key regulator in the autophagic response to oxidative damage, as HDAC6 is recruited to ubiquitinated autophagic substrates, where it stimulates autophagosome–lysosome fusion by promoting F-actin remodeling in a cortactin-dependent manner.33 However, HDAC6 and cortactin are dispensable for starvation-induced autophagy.
CMA differs from the other types of autophagy as it does not involve vesicle formation but, rather, a direct translocation of a specific set of soluble proteins across the lysosomal membrane for subsequent degradation (Fig. 24.2).34 CMA cargo substrates include enzymes, transcription factors, binding proteins, subunits of the proteasome and proteins involved in vesicular trafficking and contain a KFERQ-like motif which is recognized by the cytosolic chaperone, Hsc70. Binding of the chaperone/substrate to the cytosolic tail of lysosome-associated membrane protein type 2A (LAMP-2A) which spans the lysosomal membrane leads to translocation of the cargo across the membrane and into the lysosomal lumen for degradation.12 This pathway has been shown to be progressively ineffective with age, because of the age-related loss of LAMP-2A.35
Microautophagy involves internalization of cytosolic cargo (cytosolic proteins, glycogen, and ribosomes) through invaginations of the lysosomal membrane,36,37 which resembles the formation of multivesicular bodies (Fig. 24.2).13,34,38 Although the molecular mechanisms in mammalian cells are poorly understood, a recent study by Sahu et al. proposes that microautophagy relies on endosomal sorting complexes required for transport (ESCRT) I and III, which are necessary for the formation of the vesicles in which the cytosolic cargo is internalized.38 It appears that this pathway, like CMA, also involves Hsc70 interaction with a substrate containing a KFERQ-like motif and that mitophagy and CMA may share common upstream pathways.34,38
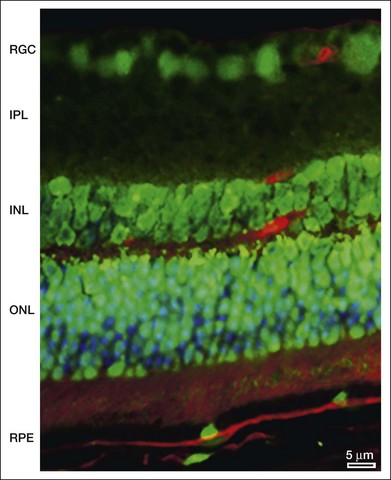
As will be discussed below, autophagy plays a critical role in maintaining retinal homeostasis as it is central, together with the proteosomal system, in the removal of damaged proteins and organelles in highly metabolic nondividing cells that exist in a pro-oxidative retinal environment. Autophagy proteins are strongly expressed in the retina (Fig. 24.4). However, problems occur when basal levels of autophagy become dysregulated as either a decrease or increase in autophagy flux will have significantly detrimental effects on cell function.39
AGE-related retinal cell loss
It is well recognized that the human retina undergoes numerous age-related changes which result in altered morphology, reduced function, and cell loss. Not surprisingly, this is associated with a significant reduction of retinal thickness as a function of age.40–42 Studies report that mean retinal thickness decreases by 0.53 µM/year40 and that, in the macula, retinal thickness and macular volume decrease by around 0.35 µM and 0.01 mm3/year.42 Changes in cell morphology include: nodular excrescences in rod outer segments43; accumulation of lipofuscin in photoreceptor inner segments and the retinal pigment epithelium (RPE)44–46; displacement of nuclei from the outer nuclear layer (ONL)47; and extension of ON-cone bipolar cell and horizontal cell processes into the ONL.48 Such evidence of retinal reorganization and plasticity has also been corroborated by animal studies.49,50 Reorganization of the dendrites could be an adaptive attempt to compensate for the lost circuitry due to photoreceptor loss and/or to make up for existing, yet dysfunctional, synapses.
There is an age-related decrease in the density of photoreceptor cells in the human retina, with rods appearing to be more vulnerable than cones.51,52 In the equatorial retina cones decrease uniformly at approximately 16 cells/mm2/year while the decrease in equatorial rods is greatest, 970 cells/mm2/year, between the second and fourth decades.52 By contrast, cone density remains relatively constant at the fovea up to the ninth decade.51–53 It therefore appears that rod photoreceptors are more vulnerable to loss during aging than cones and that photoreceptors in the fovea are less susceptible to attrition. Furthermore, compensatory adaptations have been reported following rod cell degeneration where the space vacated by dying rods is filled by enlarged rod inner segments from neighboring photoreceptors, resulting in similar rod coverage at all ages.51,54–56 Evidence that cones depend on survival factors secreted by rods may explain this differential vulnerability between rods and cones57–60 but remains a matter of intense debate.
The loss of photoreceptors appears to precede the loss of associated neural cells. The retinal nerve fiber layer thickness decreases dramatically with age40,61 and is associated with a significant loss of retinal ganglion cells (RGC) by as much as 150/mm2 over a period of 40+ years.41 RGC death at the equatorial regions follows a similar trend as the photoreceptors during aging, thus maintaining a constant photoreceptor-to-RGC ratio.52 Age-associated degeneration of the rod bipolar cells in the inner nuclear layer (INL) has been reported.62 Although rod cell death can initiate as early as the second decade of life, the bipolar cells start to degenerate only after the fourth decade and appreciably reduce by the ninth decade.52 So it seems to be a phenomenon secondary to rod cell loss. In a more comprehensive study done using multiphoton confocal microscopy to quantify neuron densities in the RGC layer, INL, and ONL, it was seen that the greatest neuronal loss occurs in the RGC layer and ONL in human aging retinas, whereas the INL is relatively preserved.63 It must also be remembered that RGCs are classified into a number of subtypes56 and so even a seemingly mild loss of RGC in the initial phases could imply the loss of a major subtype of RGC that could start affecting visual perception.
Despite numerous studies to determine age-related changes in RPE cell density, outcomes vary and are highly dependent on retinal location. Two studies have reported that RPE density decreases with age in the equatorial retina and is greatest in the periphery,52,64 with an estimated loss of 0.3% per year.64 By contrast, no significant age-related decrease in RPE cell density was observed at the foveal center, suggesting that, like foveal cones, the RPE cells in this region are more resistant to attrition than those outside the fovea.52 However, a further study reported that the macular region in aged eyes contained a significant number of apoptotic cells and these were greatest in the fovea.54 Given the disparity between studies on age-related changes in RPE cell density it is not surprising that there is no clear agreement to what extent the RPE-to-photoreceptor ratio changes with age, if at all.52,65
Evidence would suggest that age-related cell loss in the retina occurs by apoptosis since occasional apoptotic cells are observed in retinal sections plus the lack of any significant age-related inflammatory response which would occur via necrosis. The stimulus for age-related cell loss is also unclear but since the majority of cells affected are postmitotic or terminally differentiated, the accumulation of stochastic damage as occurs with aging in other tissues is plausible. In particular, oxidative damage is likely to play a significant role since the retina has high oxygen levels, is exposed to light, and has a number of highly metabolically active cell types, making it an ideal environment for the generation of reactive oxygen species.66 As already mentioned, the retina undergoes considerable remodeling throughout life to adapt to age-related cell loss. In addition, there is likely to be a basal level of limited cellular replacement through resident and bone marrow-derived stem or progenitor cells that have the capacity to differentiate into a number of different retinal cell types.67
Retinal damage: death and repair
Glaucoma and ganglion cell loss
Glaucoma is a heterogeneous group of diseases that leads to RGC death.68–70 Pathology is associated with “cupping” of the optic disc due to loss of ganglion cell axons. Death of RGCs in both postmortem specimens and experimental animal models takes place by apoptosis.71–73 Analysis of retinas from human donors suffering primary open-angle glaucoma demonstrated greater than 15 times more terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive apoptotic cells than the controls.74 Interestingly, apoptosis also accounts for the selective elimination of about 50% of excess RGCs during developmental organization of the visual pathway.75 Apoptosis of RGCs in glaucoma has generally been considered to occur as a result of mechanical damage due to an increase in intraocular pressure, but damage to RGCs can also occur in normal-pressure glaucoma.76 Other insults that have been reported to induce RGC apoptosis are neurotrophin deprivation, glial activation, ischemia, glutamate excitotoxicity, and oxidative stress.70 Confirmatory data that RGC apoptosis occurs by the intrinsic pathway that involves the mitochondria come from backcrossing DBA/2J mice that exhibit a spontaneous secondary glaucoma with Bax (one of several BH3 family death proteins) knockout mice that resulted in a mouse strain in which glaucomatous neurodegeneration was reduced.77
Despite the strong association between oxidative stress, mitochondrial damage, and RGC death, there have only been limited studies on the role of autophagy in RGC maintenance and glaucoma. Rodriguez-Muela and colleagues recently reported that autophagy promotes survival of RGCs after optic nerve axotomy in mice.78 Calpain-mediated cleavage of Beclin-1 and autophagy deregulation have been reported in a rat model of retinal ischemic injury that recapitulates features of glaucoma.79 Furthermore, blockade of autophagy increased cell death in cultured RGC, suggesting a prosurvival role of the autophagic process. By contrast, activation of autophagy in RGCs occurs after optic nerve transection and increased autophagy offers a protective role in cultured RGCs.80 Sternberg and colleagues demonstrated that autophagy provided a survival mechanism against caspase-dependent death of neonatal RGCs induced by axon damage.81
Diabetic retinopathy
It has long been recognized that diabetes leads to a loss of pericytes and endothelial cells in the retinal vasculature (Fig. 24.5).82,83 Development of new techniques to study the vasculature allowed Cogan and colleagues to identify pericyte “ghosts” (intramural pockets in the vascular basement membrane lacking normal cell contents) as one of the earliest changes in DR.84 Pericyte loss is accompanied by loss of endothelial cells from the retinal vasculature, leading to “acellular” capillaries (intact vessel basement membrane devoid of cells lining the lumen). Cell death in these populations appears to occur predominantly via the intrinsic apoptotic pathway.85 These characteristic changes have long been considered a hallmark of DR. However, studies by Barber and others reveal that diabetes is also associated with increased loss of retinal neurons.86,87 STZ-diabetic rat retinas after 30 weeks of diabetes showed a 22% reduction in the thickness of the inner plexiform layer, a 10% reduction in RGCs, and a 14% reduction in the thickness of the INL.88 Clinical studies of diabetics using scanning laser polarimetry and optical coherence tomography have largely confirmed these findings in patients.89–91 It is also likely that other neuronal populations such as amacrine cells are also undergoing apoptosis in diabetes.92 The potential initiators of retinal cell apoptosis are many and include hyperglycemia, oxidative stress, reduced blood flow, ischemia, neuroinflammation and, specifically in the case of retinal neurons, glutamate excitotoxicity.85,86
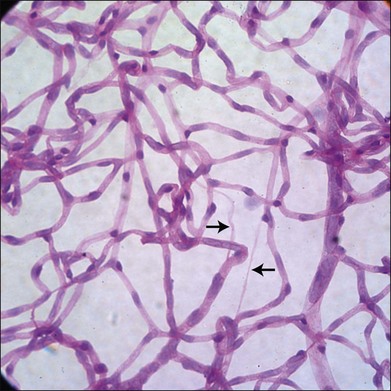
There is surprisingly little information currently available on the potential role of autophagy in the pathogenesis of DR, even though there is extensive evidence that: (1) autophagy is dysregulated in other diabetic tissues93,94 and (2) damaged mitochondria are associated with the pathogenesis of DR.85 We have recently reported that autophagy flux is decreased in the retinal cells of diabetic rats compared to controls.95
The impact of cell death in the diabetic retina and the order in which different cell types die during the progression of DR are unclear. For example, pericyte dropout and acellular capillaries are observed in many diabetic animal models, yet they do not progress to the sight-threatening proliferative stage. Furthermore, the duration of diabetes in many patients may be 15 years or more before clinical abnormalities are observed in the retina, even though vascular and neuronal cell death will be occurring. A possible explanation for this chronic, rather than acute, attrition in the retina is a low level of cellular replacement from resident and bone marrow-derived stem or progenitor cells.67 Bone marrow-derived progenitor cells have the capacity to differentiate in a range of retinal vascular and neuronal cell types in response to retinal injury and in the case of the retinal vasculature can repopulate acellular capillaries in rodent models of DR.96,97 However, these bone marrow-derived progenitors are reduced and appear to be dysfunctional in diabetics and thus repair potential is attenuated.98,99
Macular degeneration
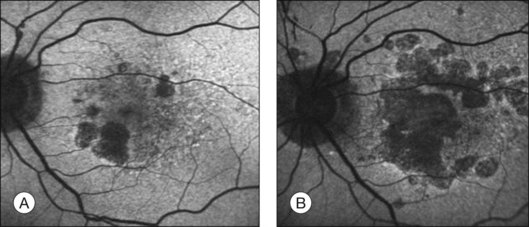
The challenge in studying retinal cell loss mechanisms in AMD is to be able to differentiate between the cell loss in the disease from that in the normal course of aging. However, reports concur that photoreceptor, RPE, and choroidal cell loss are accelerated in AMD and that this is regional and often focal. Clinically, areas of geographic atrophy can be observed within the retinal arcades in the eyes of patients with dry AMD and these lesions will increase in size with lengthening duration of AMD (Fig. 24.6). Areas of geographic atrophy show a continuous enlargement over time with a median growth rate of between 1 and 13 mm2/year and progression appears to be linear.100,101 The considerable variability between patients is unexplained but could reflect genetic susceptibility, diet, smoking, and light exposure. However, there is concordance of disease progression in bilateral geographic atrophy.102 Loss of RPE is normally associated with a reduction in the choroidal vasculature. Evidence of the degree of choroidal capillary loss and its relationship with RPE loss has been elegantly described by McLeod and colleagues.103 They observed a linear relationship between the loss of RPE and choriocapillaris in geographic atrophy. The vascular area was reduced by 50% in regions of complete RPE atrophy and there was extreme constriction of remaining viable capillaries. Adjacent to active choroidal neovascularization, choriocapillaris dropout was evident in the absence of RPE atrophy, resulting in a 50% decrease in vascular area. The authors concluded that the close association observed between degenerating RPE and choriocapillaris suggests that, at least in geographic atrophy, RPE atrophy occurs first, followed by choriocapillaris degeneration.
It has largely been considered that photoreceptor cell death occurs as a result of dysfunction or death of the underlying RPE. However, as discussed above in the aging section, there is significant rod photoreceptor loss as a function of age even in the presence of an apparently healthy RPE.51 Photoreceptor topography in both dry and wet AMD shows preferential loss of rods over cones.104 The total number of foveal cones in eyes with large drusen and basal deposits was similar to that of age-matched controls and the foveal cone mosaic appeared normal. By contrast, cones appeared large and misshapen and few rods remained in the parafovea and, by late-stage AMD, virtually all surviving photoreceptors in the macula were cones.104 In eyes from wet AMD donors, photoreceptors surviving on, or at the margins of, disciform scars were largely cones. Subsequent functional studies supported the histological evidence for preferential vulnerability of rods in aging and AMD.105 Although Jackson and colleagues concluded from these histological studies that photoreceptor loss is secondary to RPE dysfunction or death in AMD, it still remains possible that a photoreceptor abnormality could be the primary effect in AMD and that this leads to subsequent loss of the underlying RPE.
Despite the overwhelming evidence of retinal cell loss in AMD there are surprisingly few reports on the type of cell death, or the initiating insult(s), in human AMD tissue. Most studies have relied on cell culture assays and animal models with an AMD phenotype which indicate that cell death occurs primarily by apoptosis. The most detailed report is from Dunaief and colleagues who observed a statistically significant increase in TUNEL-positive apoptotic cells in the inner choroid, RPE, ONL, and INL of AMD donor eyes.106 This increase in the number of apoptotic cells was evident in 5 of 6 eyes with geographic atrophy and all eyes with exudative AMD. Interestingly, in eyes with drusen, only a few TUNEL-positive cells were observed in each nuclear layer except the RGC layer. From the same study TUNEL-positive photoreceptors are also evident over an area of disorganized RPE near an edge of atrophy. TUNEL-positive RPE cells were found most commonly near areas of atrophy and occurred more often in AMD eyes than in controls.106 While an interesting and informative study on cell death in AMD, the number of apoptotic cells seems high, especially in the neural retina, for a slowly progressive condition. By contrast, Xu et al. only observed apoptotic cells in the retina of 4 of 16 AMD donor eyes and the overall numbers of apoptotic cells appeared relatively low.107 Nordgaard and colleagues undertook proteomic analysis of the RPE from donor eyes at progressive stages of AMD.108 Several components of apoptotic signaling pathways (αA crystallin, VDAC1, HSP70, GST-π) demonstrated changes in expression early in AMD or changed linearly with AMD progression. Surprisingly, information on death mechanisms in the AMD is scarce but there is evidence from cell culture and animal models for both intrinsic and extrinsic apoptosis pathways. FAS-mediated apoptosis has been observed in retinal cells, which could explain the role of inflammation in AMD,109 and a multitude of studies have reported oxidative stress-induced apoptosis by the classic mitochondrial route.110–113 The recent finding of Alu RNA accumulation leading to RPE cytotoxicity in geographic atrophy and Dicer1 downregulation suggests a completely new mechanism of RPE degeneration.114 It should also be considered that cellular repair, cellular replacement, and damage control are critical in retinal homeostasis and a declining antioxidant system together with increased oxidative damage will play a major etiological role in AMD, and that once the threshold for damage is reached, multiple cellular processes for repair and regeneration will be impacted.115–118
There is increasing evidence that autophagy flux may be dysregulated in the RPE in AMD119,120 and is likely due to multiple factors that affect the initiation of autophagy and/or the fusion of autophagosomes with lysosomes. These will include lipofuscin accumulation, susceptibility to oxidative stress, mitochondrial damage, and lysosomal dysregulation, all of which have a strong association with AMD. To what extent changes in autophagy flux reflect alterations in the formation or elimination of autophagosomes and are a cause or consequence of AMD remains to be determined. Lipid peroxidation products reduce autophagy flux and increase lipofuscin accumulation in cultured RPE cells.121 An increase in lysosomal pH which is associated with the lipofuscin constituent A2E122 may impair autophagosome–lysosome fusion, as may the accumulation of lipofuscin granules within the lysosomal vacuome. It has been reported that drusen formation may reflect an increase in both mitochondrial damage and autophagy in the RPE.123 The researchers speculated that increased autophagy and the release of intracellular proteins via exosomes by the aged RPE may contribute to the formation of drusen. It is important to note that there is substantial cross-talk between autophagy and proteasomal degradation pathways which may also affect the status of the RPE.119
Retinal detachment
Retinal detachment resulting from full-thickness retinal breaks, subretinal exudation, and/or vitreoretinal traction is a common cause of photoreceptor loss.124 Cell death is highly dependent on the area and duration of the detachment. Analysis of tissue samples from patients with retinal detachment showed the presence of significant numbers of apoptotic cells by 24 hours, which peaked by 2 days and dropped to a low level by 7 days after detachment.125 These observations have been largely supported by experimental retinal detachment in cats and rats, both of which show a peak of apoptosis between 1 and 3 days postdetachment, which then declines.126,127 Some apoptotic cells were still evident at 28 days postdetachment. However, there is some debate whether apoptosis following retinal detachment occurs via the intrinsic or extrinsic pathways.124,128,129 Interestingly, retina RPE separation in rats causes a Fas-dependent activation of autophagy in injured photoreceptors and, if autophagy is inhibited, the time course and number of apoptotic cells are accelerated.130 Thus it appears that autophagy is activated to regulate the level of receptor apoptosis.
Retinal dystrophies
Retinal dystrophies encompass a heterogeneous group of inherited conditions, with more than 100 genes or loci identified so far.131 The most common subtype is retinitis pigmentosa (RP), which is characterized by progressive death of retinal rod and cone photoreceptors, and the disease proceeds toward reduction of peripheral field with tunnel vision and finally loss of sight.131,132 One of the major impediments in the comprehensive identification of the degenerative mechanisms of retinal dystrophies is the heterogeneity of the disease and involvement of multiple causal genes in the pathogenesis of the disease.133 The mode of rod cell death in several animal models of RP suggests death by apoptosis, which is in agreement with findings in RP retinas from donor eyes134 (reviewed by Travis135). Cone cells usually die as a secondary response to rod cell death, possibly because they depend on rod-secreted neurotrophic factors for survival.57,60
Studies of apoptotic mechanisms in the photoreceptors of RP models imply the involvement of caspase-dependent as well as independent pathways.136–138 DNA fragmentation was a regular feature encountered in the mouse models, indicating that photoreceptors in mouse models die of apoptosis. Administration of caspase-3 inhibitors inhibited photoreceptor apoptosis in the tubby mouse (Usher syndrome model).139 Caspase-independent modes of apoptosis may involve calpain and calcium ion excess in RP.140 In recent years it has been suggested that, although the primary cell death mode will be apoptosis, other modes of cell removal could be involved, including autophagy and complement-mediated lysis.141 Investigation for different cell death pathways in three independent mouse models of photoreceptor degeneration – the rd/rd mouse, the rds/rds mouse and the light-damage model in albino mice – shows that, apart from apoptotic cell death, several oxidative stress markers as well as elements of the autophagic and complement pathways are upregulated. While the induction of oxidative stress response genes is early, the induction of autophagy was only seen in damaged retinas when compared to controls, which made the authors conclude that autophagy specifically removes damaged photoreceptors from the retina.141 However, the data may also be interpreted as an attempt in the damaged retina to salvage the photoreceptors from initial stress which, when overwhelming, gives rise to autophagic death. Finally, the evidence of upregulation of high-mobility group box 1 (HMGB1) protein in human eyes with retinal detachment suggests that necrosis could also be a mode of photoreceptor death.142 It could well be this mode of cell death that accounts for some of the caspase-independent photoreceptor death pathways that have hitherto been thought to be apoptosis. However the relevance of necrosis in early stages of RP still needs to be investigated. Nevertheless, the heterogeneity of RP and similar retinal dystrophies necessitates the understanding of the earliest mechanisms of disease inception such that customized treatments may be catered according to the nature of disease pathology in a patient. Attempts to block cell death by one strategy may prove to be futile as the protective effect may only be successful for a short duration, after which the cell might proceed through another death mode.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


