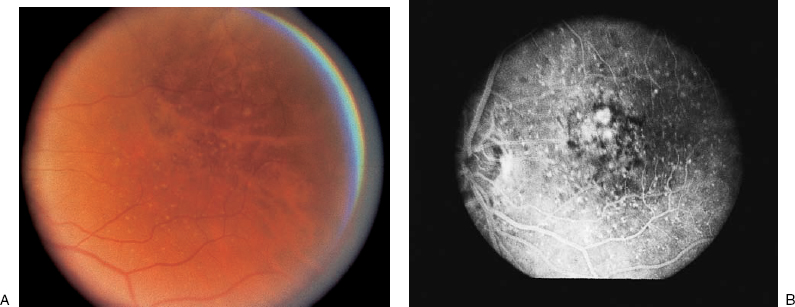
9 What are some of the disease associations with hyperpigmented lesions of the fundus? Fine, hyperpigmented lesions of the fundus may be associated with infectious and inflammatory retinal diseases, or they may exist with dystrophic and degenerative diseases of the retina. Common causes of these hyperpigmented lesions of the fundus include hyperplasia of the retinal pigment epithelium (RPE) in response to infections or inflammatory diseases (uveitis), hereditary or congenital disorders of the retina, such as rod-cone (retinitis pigmentosa), or cone–rod dystrophies (Fig. 9–1). Degenerative diseases of the retina and RPE include central or peripheral retinal degeneration and associated choroidal disease or neovascularization (CNV), which may present with fine, hyperpigmented lesions. In diseases such as age-related macular degeneration, fine, hyperpigmented lesions may be seen as a part of the disease involving the RPE-Bruch membrane complex. Peripheral retinal degenerative diseases, such as peripheral cystoid degeneration may present with areas of pigment epithelial atrophy and hypertrophy, possibly presenting as fine hyperpigmented lesions. Symptoms associated with hyperpigmented spots may include floaters if an underlying infectious or inflammatory (uveitis entities) disease of the retina and choroid is present. If these fine, brown–black spots are located centrally, that is, within the macular area, they might be associated with the complaint of a central scotoma. Disease entities such as syphilis may be associated with retinitis or vitreitis, and the patient may present with floaters or reduced vision. Any of the causes of uveitis, such as sarcoidosis or Vogt–Koyanagi–Harada, may present with floaters. The floaters associated with sarcoidosis, for example, often are associated with retinitis or an inflammatory cellular response in the vitreous. Postinflammatory changes such as fine hyperpigmented spots may be seen with many of the infections or inflammatory diseases of the retina. Floaters are less likely to be seen with hereditary, congenital dystrophic diseases or degenerative diseases of the retina. Fine, hyperpigmented spots. If these spots are located within the macular area, and associated with diseases like cone-rod dystrophy or age-related macular degeneration, can be associated with reduced color vision, reduced contrast sensitivity, scotoma, or metamorphosia. Rubella, caused by an RNA virus of the togavirus group, can be associated with congenital and acquired postnatal infections. The association between congenital rubella and infection of mothers in the first trimester of pregnancy was recognized as early as 1941.1 The virus was isolated in 1962, and a vaccine was developed in 1969; subsequently, occurrence of the virus decreased.2 Infections during the first trimester of pregnancy may result in spontaneous abortion or stillbirth. Children who acquire the virus in utero may present postnatally with a spectrum of findings known as the congenital rubella syndrome. This maternal rubella syndrome is characterized by mental retardation, congenital heartdisease, and deafness and has associated ophthalmic findings that include cataracts and anterior and posterior segment inflammation. When the infections occur postnatally or are acquired, there is a mild, self-limited illness with associated fever, adenopathy, and skin rash. The pigmented changes that occur with rubella retinitis can be fine and dust-like in character, or there may be large, discrete areas of hyperpigmentation. The pathognomonic retinal appearance and the fine, powdery granular appearance usually suggest the diagnosis of rubella retinitis. Rubella retinitis is seen in 25% or more of infants with the syndrome. Anterior-segment inflammation and vitreous inflammation can be present. Along with the granular hyperpigmented and hypopigmented changes, optic disc pallor may be present. The course of the retinitis is most often benign. Histopathologic changes can include necrosis of the iris pigment epithelium, chronic granulomatous inflammatory changes of the iris, and alternating areas of RPE hyperplasia and atrophy. Maternal immunization provides a protective effect against fetal acquired viral diseases. With maternal immunization, immunoglobulin G (IgG) antibodies are produced, cross the placenta, and provide immunization for the mother and the fetus. Immunization of pregnant women with viral vaccines for poliovirus, influenza viruses, and rubella are safe for both the mother and the fetus.3 Retinal degenerations and dystrophies such as retinitis pigmentosa are characterized by progressive loss of vision (with the time course often dependent on the genetic heterogeneity), particularly night vision. These retinal degenerations are characterized by the loss of photoreceptors or photoreceptor outer segments, particularly rod photoreceptors in the earlier course of the disease. The genetic heterogeneity of the degeneration often expresses the degree of severity of vision loss. With the prevalence of 1 in 4000 worldwide, Bundey and Crews projected the genetic incidences to be 19% autosomal dominant, 19% autosomal recessive, 8% X-linked, 8% undetermined, and 46% simplex or sporadic.4 Often retinitis pigmentosa and allied diseases have characteristic retinal, electrophysiologic, and psychophysiologic changes. The retinal examination may include vascular attenuation, intraretinal bony spicule pigmentation, and areas of RPE depigmentation throughout the midperiphery. The optic discs appear pale, macular edema may be present, and vitreous syneresis or opacities may be present. Characteristic electrophysiologic changes include subnormal to abnormal B-wave and A-wave amplitudes and prolonged B-wave implicit times, particularly in dark-adapted patients tested with blue light. The 30-Hz flicker electroretinogram (ERG), which evaluates cone system responses, is delayed in the early stages of retinitis pigmentosa. The inheritance of an X-linked trait depends on the segregation of maternal X-chromosomes. X-linked recessive inheritance is typified by the expression of traits in males; females are carriers of the X-linked traits. These disorders are expressed by all males inheriting the mutant gene but are seen only in females homozygous for the recessive allele. For some diseases such as X-linked retinitis pigmentosa, choroideremia, ocular albinism, and the mucopolysaccharidoses, the carrier state may be detected in females. One of the two X-chromosomes in each of a female’s cells is randomly inactivated by a process referred to as lyonization. If a disproportionate number of normal X-chromosomes are inactivated, mild manifestations of the disorders may occur. Carriers of these X-linked forms of disease may show a spectrum of disease dependent on the extent of inactivation of the normal X-chromosome and may show slowly progressive retinal degeneration.5 Female carriers of X-linked retinitis pigmentosa may show a patch of bony spicule pigment or may have an abnormal macular tapetal reflex. Electrophysiologic testing may be helpful in detecting female carriers of X-linked retinitis pigmentosa.5,7 This can be detected by ophthalmoscopy or full-field ERG testing or both. These females have a 50% chance of having an affected male carrier and a 50% chance of having a carrier daughter.6 X-linked inherited choroideremia presents in young men with often normal vision, increased dark-adaptation thresholds, and full visual fields to large test objects, even at the time when granularity and depigmentation of the RPE can be seen around the retinal periphery. Presenting in the first or second decade of life, these patients progressively develop problems with dark adaptation progressing to night blindness and constriction of visual field. Central vision is severely impaired by the fifth decade of life. This condition is characterized by a progressive degeneration of the RPE, retina, and choroid. Electrophysiologic responses may be affected within the first decade, and the ERG responses may be extinguished as the disease progresses. Fundus abnormalities with patchy depigmentation and stippling of the RPE may occur in affected individuals as early as the first decade of life and also can be seen in asymptomatic female carriers of choroideremia. As the disease progresses with RPE and choroidal atrophy, only small scalloped areas of intact choroid in the macula and periphery may remain. In carriers, histologic studies reveal scattered areas of reduced photoreceptors, RPE atrophy, pigment clumping, and associated areas of choriocapillaris loss.8 Choroideremia is related to a gene isolated by linkage analysis to Xq13-q22, whose protein product, Rab escort protein (REP-1), is noted to be absent in peripheral lymphocytes. The absence of REP-1 results in protein truncation either by deletions, translocations, or mutations. MacDonald developed a simple immunoblot assay using anti-REP-1 antibodies to diagnose patients with choroideremia.9 In ocular albinism, characterized by subnormal to abnormal vision and pendular nystagmus, patients may present with symptoms of photoaversion, clinical evidence of iris transillumination defects, and a hypopigmented fundus. The fovea is hypoplastic, with no foveal reflex or yellow macular pigmentation and an absence of the foveal pit on histology. True ocular albinism is divided into ocular and oculocutaneous albinism. In oculocutaneous albinism, the skin and eye may be affected; in ocular albinism, the eyes are clinically affected with some degree of cutaneous pigmentary change. Oculocutaneous albinism is inherited as an autosomal-recessive trait. Ocular albinism is transmitted as an X-linked disease and results from a reduction in the number of melanosomes. Female carriers of ocular albinism may have iris transillumination defects, macular pigment epithelial mottling, and irregular areas of patchy hypopigmentation in the midperiphery of the fundus. Female carriers of Nettleship-Falls ocular albinism (X-linked) can be identified by skin biopsy showing macromelanosomes or giant pigment granules in the RPE, the ciliary epithelium, and the keratinocytes and melanocytes of the skin. Macromelanosomes are lacking in the Forsius-Erickson type of X-linked ocular albinism.10 Genetic analysis of the ocular albinism 1 gene demonstrated seven presumed pathogenic mutations in nine families examined with X-linked ocular albinism, with five single nucleotide substitutions predicting a change in the conserved amino acids. The mutations present predict crucial changes in the protein structure that is involved. Clinical examination failed to identify any phenotype–genotype pattern.11 These cellular lysosomal storage diseases result from deficiencies of specific lysosomal exoenzymes involved in the degradation of dermatan, heparan, or keratan sulfate, inborn errors of lysosomal glycosaminoglycan metabolism. Mucopolysaccharides (MPSs) that have been incompletely degraded accumulate in organs and connective tissues and are excreted in the urine. The syndromes are varied but may produce skeletal deformities, coarse facies, visceromegaly, cardiac disease, deafness, mental retardation, and ocular involvement. Subclassification of the syndromes may occur on clinical grounds alone. The MPSs usually are transmitted as autosomal-recessive traits, except type II (Hunter), which is X-linked recessive. The MPS syndromes, particularly the autosomal-recessive syndromes associated with storage of heparan sulfate, such as MPS Hurler and MPS Scheie syndromes, may have an associated retinal dystrophy. MPS II, or Hunter syndrome, with X-linked inheritance, may demonstrate a pigmentary retinopathy. The patients may also have corneal clouding, coarse facies, dwarfism, and mental retardation. The enzymatic defect is a deficiency in iduronate sulfatase, an enzyme needed in the sequential degradation of heparan sulfate and dermatan sulfate. The Hunter syndrome is further divided into type A and type B. Type A Hunter syndrome includes patients with symptoms by age 1, who die early. MPS II-B begins at about age 4 years. These patients have less severe symptoms, and may live into their sixties. Ophthalmic manifestations of MPS II include progressive retinal dystrophy and papilledema. The ERG may be reduced or extinguished. Night blindness and continued deterioration of vision may be common. Bony spicule pigmentation and pigment epithelial atrophy may be noted in the midperipheral fundus. Not all patients show elevated glycosaminoglycan levels in the urine, and enzyme assays with evaluation of the electrophoretic patterns of the affected glycosaminoglycans (GAGs) become necessary. Carriers can be identified by the quantitation of fructose-1 phospate in fibroblast cultures, determination of iduronate sulfatase activity in hair roots, and enzymatic assays of GAGs.12 Since the discovery of penicillin in 1943,13 the incidence in syphilis has decreased, particularly in the 1970s, with this specific therapy; however, there has been a resurgence in incidence since the 1990s along with the increased incidence of human immunodeficiency virus (HIV). Following the initial infection, there is a humoral and cellular response. Clinical manifestations include primary, secondary, latent, tertiary syphilis, cardiovascular syphilis, and neurosyphilis in acquired disease. Congenital syphilis results from transplacental transmission of primary or secondary syphilis. Ocular manifestations of secondary and tertiary syphilis include the following anterior segment changes: conjunctivitis, keratitis, iris nodules, iridocyclitis, episcleritis, and scleritis. Posterior segment changes include retinitis, choroiditis, vasculitis, disc edema, vasculitis, neuroretinitis, choroiditis, choroidal effusion, optic neuritis, and exudative retinal detachments. Choroidal neovascular membranes have been reported. Healed areas of neuroretinitis, particularly in the peripapillary region, may result in a pigmentary retinopathy similar in appearance to retinitis pigmentosa.14,15 Secondary syphilis, occurring 6 weeks to 6 months after primary infection, may be associated with a granulomatous or nongranulomatous anterior uveitis and vascularized iris papules. Following the posterior segment uveal involvement, the patient may have extensive gliosis and atrophy of the RPE in the posterior pole. With the increase in HIV during the early to mid-1990s, there have been reports of syphilis occurring concurrently in HIV-positive patients. These patients may present with uncharacteristic clinical manifestations of syphilis. In addition, acute retinal necrosis and acute retinitis have been associated with syphilis in patients coinfected with HIV.16 Regression of these areas of retinitis may lead to small, hyperpigmented spots in the retina. Congenital syphilis is characterized by keratouveitis from acute interstitial keratitis occurring in patients aged between 5 and 25 years and later will result in interstitial keratitis. Congenital syphilis may also result in a bilateral salt-and-pepper fundus that may occur throughout the fundus. There is no progression of the retinopathy and little effect on the visual acuity. Ocular syphilis is often referred to as the “great masquerader.” Its presentation with anterior and posterior granulomatous or nongranulomatous uveitis or diffuse inflammation of the anterior segment, posterior segment, and optic nerve often allows the presentation to include the differential diagnosis of sarcoidosis, sympathetic ophthalmia, Vogt–Koyanagi–Harada syndrome, Behçet syndrome, acute retinal necrosis syndrome associated with herpes family viruses, toxoplasmosis, and other causes of acute anterior and posterior uveitis. Serologic screening tests include the rapid plasma reagent (RPR) or the Venereal Disease Research Laboratory (VDRL) test. Infection with Treponema pallidum stimulates antibodies against cardiolipin. The VDRL test may become negative, but it is usually positive in patients with primary and secondary syphilis. The VDRL test may become negative later in the disease, particularly tertiary disease, and becomes negative after treatment. More specific tests to confirm infection include the fluorescein treponema antibody absorption (FTA-ABS) test, which is more sensitive than the VDRL test at all stages of the syphilitic infection. Primary and early secondary syphilis is treated with 2.4 million units intramuscularly; daily for 10 days. Most authorities believe that tertiary and ocular syphilis and neurosyphilis should be treated with 2–4 million units of intravenous penicillin every 4 hours for 10 days to 2 weeks. Patients allergic to penicillin are treated with doxycycline or erythromycin. As the population over the age of 60 years continues to increase, age-related macular degeneration (AMD, ARMD, or ARM) will continue to be the leading cause of irreversible blindness in developed countries.17 AMD is classified as the exudative type associated with choroidal neovascular membranes, occurring in 10 to 15% of cases, and the nonexudative, or dry type, occurring in 85 to 90% of cases and characterized by abnormalities of the RPE with atrophy or hyperplasia and drusen. Changes in macular pigmentation can be found in both forms of AMD. To standardize communications about AMD, an international classification and grading system has been suggested. Early age- related maculopathy (ARM) in this classification is characterized by the presence of drusen and RPE abnormalities. Drusen, subretinal, yellow deposits, may vary in size. Hard drusen are <63 μm and soft drusen are 125 μm. Large drusen may become confluent and coalesce into drusenoid RPE detachments. These areas may progress to areas of geographic atrophy or exudative disease. Late ARM is characterized by geographic atrophy of the RPE and neovascular disease. Late AMD is the most likely to be associated with severe visual loss.18 Age-related macular degeneration is the leading cause of severe central visual acuity loss in people over 65 years of age in the United States and in other white populations. Persons over the age of 75 have a 30% chance of manifesting some of the early forms of AMD with drusen and RPE abnormalities. Of those remaining, 23% will have evidence of AMD over the subsequent 5-year period.19,20 In a population of 3583 adults, Klein and colleagues studied the incidence and progression of retinal drusen and retinal pigmentary abnormalities and the signs of late AMD. Persons over the age of 75 years, compared with those between 43 and 54 years of age, had a higher 5-year incidence of the following: (1) larger drusen (125 to 249 μ) in 17.6% versus 2.1%; (2) soft drusen in 16.3% versus 1.8%; and (3) RPE abnormalities in 12.9% versus 0.9%, exudative macular degeneration in 1.8% versus 0.07%, and geographic atrophy in 1.7% versus 0%. Late AMD was more likely to develop in eyes with soft, indistinct drusen (6.5% versus 0.1%) or RPE abnormalities (7.1% versus 0.1%).19 A meta-analysis of the Beaver Dam Eye Study, the Rotterdam Study, and the Blue Mountain Eye Study (14,752 participants with gradable photos) indicates that there is 0.2% prevalence of AMD in patients between ages 55 to 64 years; this prevalence increases to 13% in the population over the age of 85 years.21 Micronutrients such as zinc, vitamin C and E, and beta-carotene have been proposed to reduce vision loss from AMD because of their antioxidant potential. These antioxidants were suggested based on the theory that oxygen free radicals liberated by light exposure might cause damage to the pigment epithelium and outer retina.22 Micronutrients such as zinc and selenium, cofactors that facilitate antioxidant enzymes, have also been considered as micronutrient supplements. Until recently, studies had failed to give convincing evidence that dietary modification or micronutrient supplementation would alter the course of nonneovascular AMD.23 In groups who are at risk for developing early (ages 40 to 59 years) or late (ages 60 to 79 years) ARM, higher levels of lutein and zeaxanthin in the diet were related to lower odds for pigmentary abnormalities, one sign of early AMD and late AMD, after adjustment for age, gender, alcohol use, hypertension, smoking, and body mass index.24 Results from the Age-Related Eye Disease Study (AREDS) support micronutrients to reduce progression of disease and vision loss in AMD. Although there is an association between ultraviolet light exposure and development of cataracts,25 clinical studies, such as the Beaver Dam Eye Study26 and the Chesapeake Bay Waterman Study,27 to date have not supported the association between light exposure and the risk of developing AMD. Reevaluation of the Beaver Dam Eye Study, a longitudinal, population-based study, found that leisure time spent outdoors increased the risk of early ARM. Participants with red or blond hair were slightly more likely to develop early ARM than were people with dark hair. Exposure to sunlight may be associated with the development of early ARM.28 Epidemiologic studies have identified increasing age, not gender, as a risk factor for AMD.19,20 Some epidemiologic studies have implicated smoking, hypertension, and other cardiovascular risk factors in the occurrence of AMD.29–31 The age-related macular degeneration risk factors study, a case-control study for risk factors of neovascular and non-neovascular macular degeneration evaluated 1222 patient sets of available photographs, and concluded that neovascular AMD was associated with moderate to severe hypertension, and particularly among patients receiving antihypertensive treatment. The group promoted the hypotheses that neovascular and nonneovascular AMD may have a different pathogenesis and that neovascular AMD and hypertensive disease may have a similar underlying cause and effect.32 Drusen and RPE changes are also risk factors for further development of AMD19–21 (Fig. 9–2). Sunlight or ultraviolet light may be a risk factor, particularly in patients with red or blond hair.28 Frank and colleagues, in reexamining the relationships between race, iris color, and AMD in a series of 306 sequential patients 60 years of age or older, confirmed the previous findings of a higher prevalence of AMD in white persons than in black persons. White subjects with light-colored irides have a higher prevalence of AMD than do those with dark-colored irides.33 The prevalence of the exudative type of AMD is higher among first-degree relatives with exudative disease. Other possible risk factors may include genetic heritage. More studies are needed to study the genetic susceptibility of AMD.34 Except for the association of beer drinking with retinal drusen in men, consumption of alcoholic beverages is not likely to be an important risk factor for the incidence of AMD.35 FIGURE 9–2. Color photograph of retinal pigment epithelium atrophy and soft drusen, risk factors for age-related macular degeneration. Age-related macular degeneration is classified a nonneovascular (or dry macular degeneration) (Fig. 9–3A) and exudative-neovascular degeneration (also called the wet form of macular degeneration) (Fig. 9–3B). Each form has its own hallmark characteristics. Loss of the RPE, choriocapillaris, and choroidal atrophy, are associated with loss of photoreceptors. Nonneovascular AMD is characterized by RPE changes, drusen, and in the late stages visual loss. Neovascular AMD may have all the characteristics of dry AMD; however, its hallmark characteristic is CNV. In 1995, the International Age-Related Maculopathy Study Group outlined a classification and grading system of clinical features of AMD, which allows more meaningful comparison of other AMD studies.18 Nonneovascular AMD is diagnosed when patients present with areas of change within the RPE or with evidence of drusen. Drusen are deposits beneath the RPE that are characteristic of but not uniquely associated with AMD. AMD is associated with two types of drusen, with different appearances and likely different prognoses. Hard drusen, less than 63 μ in diameter, have distinct borders. They appear as yellow, small nodules beneath the RPE and can precede the development of atrophic AMD. Soft drusen appear large (usually larger than 125 μm in diameter) and pale yellow or grayish white. They may be dome-shaped elevations that may resemble localized serous RPE detachments. As drusen disappear, areolar atrophy may be seen. Spontaneous drusen regression probably involves RPE atrophy.36 Clinicopathologically drusen appear to be either focal lipoidal degeneration of RPE cells or localized accumulation of hyaline material in the inner and outer collagenous zones of a normal Bruch’s membrane.37 In this study, Green and Enger observed few RPE degenerative changes with hard drusen. During fluorescein angiography (FA), hard drusen hyperfluoresce. Histopathologically, soft drusen represent localized detachments of the RPE. Drusen may be associated with basal linear or basal laminar deposits. Basal linear deposits refer to material located external to the RPE basement membrane, lying within the inner collagenous zone of the Bruch’s membrane. These deposits consist of granular and vesicular lipid-rich material with wide-spaced collagen. Basal linear deposits are commonly seen histologically in eyes with vision loss and are associated with geographic atrophy or subretinal neovascular membranes. In contrast, basal laminar deposits are found between the plasma membrane and the basement membrane of the RPE. On electron microscopy, these lesions consist mainly of wide-spaced collagen and electron-dense bodies. Soft drusen are small pigment epithelial detachments which can appear hypofluorescent or hyperfluorescent on FA. Soft drusen are believed to be a hallmark of AMD, associated with abnormalities of the RPE-Bruch’s membrane complex. The development of CNV and geographic atrophy of the RPE appears to be associated more frequently with soft drusen than with hard drusen. FIGURE 9–3. (A). A fluorescein angiogram showing hyperfluorescence in an area of retinal pigment epithelium atrophy, a “window defect.” (Reproduced with permission of Novartis Ophthalmics, Inc.) (B). A fluorescein angiogram of a patient with hyperfluorescence and leakage associated with a choroidal neovascular membrane. (From Ophthalmologica. 2001;215:247–253, reproduced by permission from S. Karger AG). Spraul and colleagues performed a histopathologic and morphometric study aimed to investigate the correlation among characteristics of choroidal vessels, the RPE, and Bruch’s membrane and the types of AMD present. Characteristics of nonexudative and exudative macular degeneration were evaluated. The nonneovascular components that were evaluated in both the macular and the extramacular regions included the degree of calcification of the Bruch’s membrane, fragmentation of the Bruch’s membrane, the number and types of drusen, basal laminar deposit, and seven morphometric variables of the choroid. Surgically excised subfoveal membranes also were processed. The observations included no statistically significant differences between eyes with neovascular and nonneovascular AMD. The single most important difference between eyes was the amount of basal laminar deposit. Eyes with AMD had significantly more basal laminar deposit in the macular area than controls as well as soft, diffuse, and large drusen. Eyes with AMD displayed fewer large choroidal vessels in the submacular choroid than did eyes without AMD. The submacular choriocapillaris density was higher in eyes with AMD. The peripheral choriocapillaris displayed the same pattern as the macular choriocapillaris. Compared with controls, a statistically significant difference was observed in the degree of calcification and fragmentation of Bruch’s membrane in eyes with exudative AMD. These differences did not reach statistical significance in eyes with nonexudative AMD. This morphometric and histopathologic study concluded that AMD is a dynamic process with early proliferation and subsequent atrophy of capillaries of the choriocapillaris. Calcification and fragmentation of Bruch’s membrane; soft, diffuse, and large drusen; and basal laminar deposit, but not hard drusen, strongly correlate with the histologic presence of AMD. The degree of calcification and fragmentation of Bruch’s membrane was more prominent in exudative AMD. These investigators concluded that the formation of choroidal neovascular membranes represents a stereotypic, nonspecific wound repair response independent of the underlying disease.38 Abnormalities of the RPE represent another form of nonneovascular AMD, including atrophy or hyperpigmentation. These changes may present as fine, hyperpigmented lesions of the fundus. Geographic atrophy of the RPE may take the form of a perifoveal, reticulated, hyperpigmented, or hypopigmented appearance; these areas enlarge and coalesce slowly over time. Large, soft, confluent drusen can develop into geographic atrophy (GAP).39 Clumps of pigmented cells at the level of the RPE may appear as focal areas of hyperpigmentation. These focal clumps of pigment may correlate with an increased risk of the development of a choroidal neovascular membrane or RPE atrophy.40 Patients with GAP may witness a significant decline in vision over time. Areas of atrophy continue to enlarge, even when they are large at baseline. This combination of decreased vision and continued enlargement of the area of atrophy, occurring bilaterally in most patients, may lead to a significant loss of vision. Geographic atrophy is the advanced form of atrophic AMD and is present in 3.5% of people aged 75 and older in the United States. Progression over time, even with sparing of the fovea until late in the course of the disease, is associated with a decrease in vision. Of eyes with geographic atrophy and good visual acuity at baseline, 40 to 50% lose three or more lines of acuity by 2 years, and 27% become worse than 20/200 by 4 years.41 Patients with GAP may develop CNV. An eye with GAP whose fellow eye has CNV is at significant risk for the development of CNV in the GAP eye. A patient with bilateral GAP and no evidence of CNV is at relatively low risk for developing CNV.42 Loss of RPE in areas of areolar and geographic atrophy may have associated loss of photoreceptors. The remaining photoreceptors may be abnormal. The outer nuclear layer disappears so that the outer plexiform layer is thinned and vacuolated with less effect on the inner nuclear layer. The cellular and molecular mechanisms underlying the death of photoreceptors and other retinal cells in AMD remain poorly understood. Patients with nonneovascular AMD presenting with drusen, particularly extensive small drusen (<63 microns), pigment abnormalities, intermediate size drusen (>63 microns), or large drusen (>125 microns), and geographic atrophy not involving the center of the macula, should be warned about the presence of macular degeneration and be advised of the need for continued monitoring with dilated examination and the use of the Amsler grid for daily monitoring. In patients with complaints of vision change, acute or chronic, fluorescein angiography is helpful in assessing changes in the pigment epithelium or evidence of CNV. Antioxidant vitamins plus zinc are now being recommended for some patients with nonneovascular AMD. The Age-Related Eye Disease Study, in a randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss, has indicated that persons older than 55 years of age should have dilated eye exams to determine the risk of developing advanced AMD. In this randomized, 11-center double-masked trial, 4757 participants were enrolled if they had extensive small drusen, intermediate or large drusen, noncentral geographic atrophy (GA), or pigment abnormalities in at least one eye, or advanced AMD, or vision loss due to AMD in 1 eye. In Category 1, defined as participants with few if any drusen, only 5 of these participants developed advanced AMD and the effects of antioxidant could not be assessed in this group. The report focused on 3640 participants. Of those enrolled, 1063 had extensive small drusen, pigment abnormalities, or at least 1 intermediate druse (Category 2); 1621 had extensive intermediate drusen, GA not involving the center of the macula, or at least 1 large druse (Category 3); and 956 had advanced AMD (GA involving the center of the macula, non-drusenoid retinal pigment epithelial detachment, serous or hemorrhagic retinal detachment, hemorrhage under the retina or the RPE, and/or subretinal fibrosis) or visual acuity less than 20/32 due to AMD in 1 eye (Category 4). Photographic assessment was used to evaluate the enrolled study participants, aged 55 to 80 years. The average follow-up was 6.3 years, with 2.4% lost to follow-up. Participants, with at least 1 eye with best-corrected vision of 20/32, were randomly assigned to receive daily oral doses containing one of the following: antioxidants (vitamin C, 500 mg; vitamin E, 400 IU; and beta carotene, 15 mg); zinc, 80 mg, as zinc oxide and copper, 2 mg, as cupric oxide; antioxidants plus zinc; or placebo. Comparison with placebo demonstrated a statistically significant odds reduction for the development of advanced AMD with antioxidants plus zinc. Participants with extensive small drusen, nonextensive intermediate size drusen, or pigment abnormalities had only a 1.3% 5-year probability of progression to advanced AMD and were excluded from the odds ratio determination for progression. The study demonstrated little or no benefit of the study formulations for persons in Categories 1 or 2. Although both zinc and antioxidants plus zinc significantly reduce the odds of developing advanced AMD (Categories 3 and 4), the only statistically significant reduction in rates of at least moderate visual acuity loss (approximately 25%) occurred in persons assigned to antioxidants plus zinc. This study has concluded that persons older than 55 years with extensive intermediate size drusen, at least 1 large druse, noncentral geographic atrophy in at least one eye, or advanced AMD or vision loss due to AMD in one eye, and without contraindications, such as smoking, should consider taking a supplement of antioxidants plus zinc such as those outlined in the study. Laser photocoagulation of drusen, to date used in nonneovascular AMD for the treatment of drusen, is one of the approaches being investigated for the management of drusen. Studies are being approached in the management of nonneovascular AMD to try to prevent the risk of visual loss and the development of CNV or pigment atrophy. Drusen may disappear from the RPE after focal laser to drusen; this disappearance may be related to the stimulation of macrophages.45 Drusen also may disappear over time without treatment.46 The Choroidal Neovascularization Prevention Trial (CNVPT), a recent randomized clinical, trial showed that light laser photocoagulation to fellow eyes of patients with CNV may increase the short-term incidence of CNV in the treated eye.47 More long-term results, with the main outcome the measure of the change in visual acuity, indicate that laser-treated eyes with a reduction of 50% or more in drusen at 1 year had more one- and two-line increases in visual acuity and less loss of visual acuity compared with laser-treated eyes with less drusen reduction or with untreated (observed only) eyes. In this study, laser-induced drusen reduction was associated with improved visual acuity and contrast sensitivity in eyes at 1 year. Long-term effects of laser-induced drusen reduction on visual function require additional observation. The overall potential value of laser treatment in eyes with high-risk drusen requires consideration of not only short-term effects on vision but also the effect of CNV and atrophy on vision and the usefulness of this procedure probably will be reassessed at a later date.48 Whereas nonneovascular AMD is characterized by drusen, RPE, and changes in Bruch’s membrane, the hallmark of exudative AMD (Fig. 9–3B) is CNV. This fibrovascular tissue, which originates from the choroidal circulation, is the leading cause of severe vision loss (SVL) in persons over the age of 65 years.49 Although it accounts for only 8% of cases of AMD, this tissue is responsible for 85% of cases of SVL associated with AMD.20 Exudative lesions may present as fine, hyperpigmented spots of the fundus if they are located in the subretinal space and associated with hemorrhage or retinal elevation. Histopathologic studies suggest that diffuse thickening of the inner aspect of Bruch’s membrane, clinically associated with large, soft drusen, predisposes Bruch’s membrane to develop cracks through which ingrowth of fibrovascular tissue from the choriocapillaris can occur. These breaks, however, can be present in eyes not presenting with CNV.50 Morphometric analysis of the choroid, Bruch’s membrane, and the RPE in postmortem eyes with AMD indicates that the single most important difference between eyes with and without AMD is the amount of basal laminar deposit. A statistically significant difference has been observed in the degree of calcification and fragmentation of Bruch’s membrane in eyes with exudative AMD compared with controls. The researchers concluded that AMD can be interpreted as a dynamic process with early proliferation and subsequent atrophy of capillaries of the choriocapillaris. Calcification and fragmentation of Bruch’s membrane; soft, diffuse, and large drusen; and basal laminar deposit, but not hard drusen, strongly correlated with the histologic presence of AMD. Calcification and fragmentation of Bruch’s membrane were prominent in eyes with exudative AMD. The formation of CNV membranes represents a stereotypic, nonspecific wound repair response independent of the underlying disease.38 Some investigators believe that abnormalities of the extracellular matrix of RPE cells may promote a proangiogenic RPE phenotype that contributes to the development of CNV.51 Accumulating evidence may suggest that endothelial growth factor (specifically vascular endothelial growth factor, or VEGF) may be implicated in chorioretinal angiogenesis. In studying the effect of VEGF on the experimental formation of CNV in rats, Honda and colleagues indicated that VEGF plays a crucial role in the formation of experimental subretinal neovascularization (SRN) and VEGF-soluble receptor gene transfection. Producing antibodies to VEGF inhibited the growth of CNV. Further studies in humans are projected, and future gene therapy for CNV in AMD is believed to be possible.52 In experimental laser-induced CNV, there is upregulation of VEGF expression, where it may be involved in promoting choroidal angiogenesis. In rats, macrophages may be a main source of VEGF in the early stage of the disease.53 To study the potential angiogenic role of macrophages in the formation of choroidal neovascular membranes, Oh and colleagues investigated the distribution of inflammatory mediators such as interleukin (IL)-1-beta and tumor necrosis factor (TNF)-alpha and angiogenic cytokines such as VEGF to identify their cellular source in surgically excised choroidal neovascular membranes (CNVMs) of various origins. Eleven surgically excised CNVMs were studied using immunoperoxidase staining to identify cellular distribution and localization of cytokines. Cytokeratin-positive cells were detected in the RPE layer, in stromal cells, and around neovascular vessels. Macrophages had similar cytokeratin-positive cells. The neovascular vessels present were immunoreactive to IL-1-beta and TNF-alpha. It was believed that the IL-1-beta and TNF-alpha secreted by macrophages may promote angiogenesis in CNVMs by stimulating VEGF production in RPE cells.54 The development of CNV is likely multifactorial. This diffuse process involves the RPE, the photoreceptor cell layer, the choriocapillaris and can involve growth factors including vascular endothelial growth factor. The early morphologic change with the development of basal deposits is not visible ophthalmoscopically, but psychophysical testing may demonstrate reduced function. The secondary changes in the pigment epithelium, soft drusen, and CNV are visible. The CNV might represent reparative responses that result in disciform scars.55 CNV may develop in eyes with GAP. An eye with GAP whose fellow eye has CNV is at significant risk for the development of CNV in the GAP eye. A patient with bilateral GAP and no evidence of CNV is at a relatively low risk for developing CNV.56 The morphologic features of angiogenesis reveal endothelial cell budding, pericyte enlargement, endothelial cell sprout formation, and development of intrachoroidal new vessels in preexisting choroidal capillaries. In one pathologic specimen, endothelial cell sprout formation was seen continuous with an intrachoroidal vessel that penetrated Bruch’s membrane. Examination of early sub-RPE new vessels show them to spread between the inner layers of Bruch’s membrane within the space usually occupied by the basal linear deposit and drusen. This ultrastructural study described two phases of new vessel growth associated with the onset of CNV with AMD. The first phase, the intrachoroidal phase, appears to be a “low-turnover” form of neovascularization that may lead to new vessels penetrating Bruch’s membrane. Extensive sub- RPE neovascularization, however, results from a “high-turnover” form of neovascularization characterized by extensive endothelial cell proliferation and migration, with each form characterized by the dynamics of angiogenesis.57 Multiple avenues are being investigated to inhibit CNV, including the use of oral protein kinase inhibitors,58,59 corticosteroids,60 and somatostatin analogs as possible effective therapies for CNV.51 The risk factors for exudative AMD and nonexudative AMD include the presence of focal hyperpigmentation and large drusen in the affected eye or fellow eye. Patients may complain of blurred or decreased vision and distortion or metamorphopsia. They may describe scotomas. Symptoms are often present when there is accumulated subretinal fluid or blood. Indirect ophthalmoscopy or biomicroscopy may reveal retinal elevation from subretinal blood or fluid or sub-RPE fluid, blood, or fibrovascular tissue, all of which may present as small, hyperpigmented, brown–black lesions of the posterior pole (Fig. 9–4). Slit-lamp biomicroscopy with a contact lens may reveal subtle subretinal fluid. Symptomatic patients should undergo FA to detect CNV. Good stereoscopic early, middle, and late-phase angiographic frames help to determine the CNV membrane boundaries. Fluorescein angiograms being used for possible treatment of CNV should be no more than 96 hours old. The Macular Photocoagulation Study62–69,70–82 helped to define the different types of CNVs by FA. The main types are classic CNVs and occult CNVs. The type of neovascularization portends prognosis and therapy recommendations. Classic CNV is seen as a well-demarcated area of hyperfluorescence on the early transit phase of the angiogram (Fig. 9–5A). There is significant leakage apparent in the later-phase frames of the angiogram as the dye pools in the subretinal space and obscures the boundary of the initially well-demarcated area of hyperfluorescence. A minority of these classic membranes may have a lacy appearance.
Brown–Black Lesions: Fine Hyperpigmented Spots
Hyperpigmented Spots
What Are Symptoms of Fine, Hyperpigmented Spots?
Rubella
Congenital and Acquired Rubella
What Is Congenital Rubella?
What Is Acquired Rubella?
What Are the Characteristics of Rubella Retinopathy?
Carrier States Associated with Fine, Hyperpigmented Spots
What Are the Symptoms and Signs of the Carrier States in Choroideremia and X-linked Retinitis Pigmentosa?
X-LINKED RETINITIS PIGMENTOSA
Choroideremia
Ocular Albinism
Mucopolysaccharidoses
Syphilis
WHAT ARE THE SIGNS AND SYMPTOMS OF SYPHILIS?
CAN SYPHILIS OCCUR CONCURRENTLY WITH OTHER INFECTIONS?
WHAT ARE THE CHARACTERISTICS OF CONGENITAL SYPHILIS?
WHAT ARE THE CHARACTERISTICS OF ACQUIRED SYPHILIS?
WHAT ARE THE SEROLOGIC TESTS FOR SYPHILIS?
WHAT IS THE TREATMENT OF SYPHILIS?
Age-Related Macular Degeneration (Age-Related Maculopathy)
WHAT IS THE BACKGROUND AND EPIDEMIOLOGY OF AGE-RELATED MACULAR DEGENERATION?
WHAT IS THE CURRENT THOUGHT ON THE USE OF MICRONUTRIENTS IN THE TREATMENT OF NONNEOVASCULAR AGE-RELATED MACULAR DEGENERATION?
DOES EXPOSURE TO SUNLIGHT CAUSE MACULAR DEGENERATION?
WHAT ARE THE RISK FACTORS FOR AGE-RELATED MACULAR DEGENERATION?
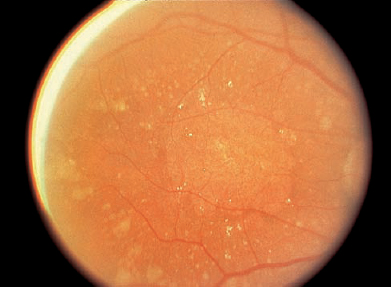
WHAT ARE THE TYPES OF AMD?
What Are Drusen and Their Characteristics?
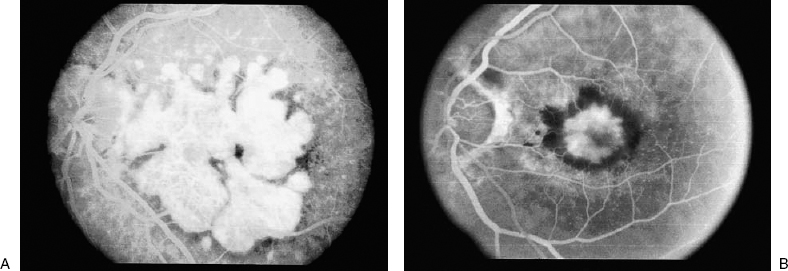
WHAT ARE THE ABNORMALITIES OF THE RPE IN NONNEOVASCULAR AGE-RELATED MACULAR DEGENERATION?
WHEN SHOULD PATIENTS WITH NONNEOVASCULAR AMD BE WATCHED ROUTINELY FOR THE DEVELOPMENT OF NEOVASCULAR AGE-RELATED MACULAR DEGENERATION?
ARE THERE ANY TREATMENTS, EXPERIMENTAL OR OTHERWISE, THAT ARE CURRENTLY BEING USED FOR NONNEOVASCULAR AGE-RELATED MACULAR DEGENERATION?
Neovascular Age-Related Macular Degeneration
What Are the Risk Factors for AMD?
What Are the Symptoms of Exudative AMD or CNV?
What Are Some of the Clinical Features of Exudative Age-Related Macular Degeneration or Choroidal Neovascularization?
What Are the Types of Choroidal Neovascularization?
Ento Key
Fastest Otolaryngology & Ophthalmology Insight Engine