CHAPTER 76 Biology of Head and Neck Cancer
Background
There are more than 40,000 new cases of head and neck cancer every year in the United States, and 500,000 worldwide, predominantly consisting of squamous cell carcinoma of the upper aerodigestive tract.1 Despite aggressive multidisciplinary treatment approaches, including preoperative or postoperative chemotherapy, radiotherapy, or both, with reconstructive surgery, there has been only modest improvement in outcomes over the past 20 years. Treatment failures still occur in the form of locoregional recurrence, distant metastases, and second primary tumors. Further insight into the molecular basis of head and neck cancer in general could lead to advances in screening, diagnosis, and treatment with improved clinical outcome.
The mechanisms underlying carcinogenesis in the head and neck are similar to those mechanisms and principles found in other solid tumors. It is generally accepted that cancer arises from the progressive accumulation of genetic alterations that lead to a selection of a clonal population of transformed cells.2 In an attempt to classify and understand the biology of sporadic and hereditary retinoblastoma, Knudson3 first put forth his model of the “two-hit” hypothesis in 1971. He described a model in which two copies of the parentally inherited Rb gene were inactivated either by mutation or by loss of chromosomal material, leading to development of hereditary or sporadic retinoblastoma. A model of tumor progression was synthesized by Fearon and Vogelstein2 into the following three basic tenets:
In 1993, Renan4 put forth a statistical model suggesting that between 6 and 10 independent genetic alterations need to accumulate before the development of HNSCC. Although the precise number of events required for HNSCC carcinogenesis is unlikely to be defined, the conclusion from this analysis is that HNSCC requires more genetic alterations in its development when compared to other solid tumors. This series of genetic changes that must occur for frank malignancy to arise is a result of the 20- to 25-year latency period seen in head and neck cancer. During this time, a cumulative exposure to carcinogens—particularly tobacco and alcohol—is believed to be the predominant cause of specific genetic alterations acquired during HNSCC progression.
Through the examination of premalignant and invasive lesions, many of these initial genetic alterations have been well described in head and neck cancer, and a genetic progression model has been created to detail the evolution of genetic alterations.5 Using microsatellite analysis, Califano and associates5 were able to correlate allelic imbalance due to chromosomal loss and gain with varying grades of dysplasia in premalignant lesions. The histologic progression of hyperplasia, dysplasia (mild, moderate, and severe), carcinoma in situ, and, finally, invasive carcinoma provided the structure in which genetic alterations were framed. A genetic progression model was created to examine lesions in each of these categories for similar patterns of allelic imbalance.5 Even in the earliest of lesions, there were clonal, genetic changes; 30% of benign hyperplastic lesions had loss at 9p21 or 3p, indicating that these losses and the corresponding inactivation of p16 and p14/aRF and putative 3p TSGs are among the first genetic alterations to occur in the progression to cancer. In addition, a transcriptional progression model has been described.6 These genetic and transcriptional alterations have been correlated with the histopathologic stages of premalignant and malignant mucosal lesions to define the order and time course of molecular alterations in the development of HNSCC.
Later, however, it was found that not just the tumor but the entire field of mucosa undergoes these changes. Slaughter and colleagues7 first coined the term field cancerization in a histologic study examining the epithelium surrounding invasive cancers. They recognized there were histopathologic changes in the epithelia surrounding invasive oral tumors and reported an increased incidence of second primary tumors, usually close to the initial tumor. Other investigators subsequently confirmed a higher incidence of second primary tumors in the upper aerodigestive tract in those patients with initial head and neck primary tumors.8–10
Thus, field cancerization originally was a clinical concept describing a mucosal surface in the region near an invasive tumor with premalignant changes and the propensity for second primary tumor development in adjacent mucosa. With the advent of contemporary molecular genetic understanding of malignancy, two theories provide possible explanations for this finding; they are (1) the areas of abnormality represent independent clones, with a unique pattern of genetic alterations, and (2) these areas of abnormality are genetically related and originate from a common cellular clone. Although these theories are not exclusive, it was shown that there was indeed lateral spread of progenitor clones throughout squamous mucosal surfaces rather than independent clones. Further studies looking at second primary head and neck tumors found that these second lesions tended to originate from a common clone.11 Another study examining serial biopsy specimens of head and neck premalignant lesions that ultimately progressed to invasive cancer showed a pattern of accumulated microsatellite loss, with the cancer demonstrating losses identical to those found in the previous premalignant lesions.5,12 Therefore, the theories of field cancerization and the genetic progression model for cancer work hand in hand to describe how the molecular and histopathologic changes occur and lead to malignancy.
Environmental Exposures and Risk Factors
The primary risk factors for the development of HNSCC are two environmental toxins, tobacco and alcohol (tobacco exposure being a much higher risk). These two factors combine synergistically to increase the risk of HNSCC. In addition to studying the mechanisms by which these toxins can alter cellular proliferation and death, investigations into the endogenous enzymatic systems dedicated to metabolize these toxins into inert molecules have been performed and are discussed. A third risk factor, which acts independently of the other two, is a history of an oral human papillomavirus (HPV) infection,13 which is addressed in more detail later in the chapter. D’Souza and coworkers13 discovered that oral HPV infection increased the risk for oropharyngeal cancer. Oropharyngeal HPV infections were detected by multiplex polymerase-chain-reaction analysis of saline rinses after cytology brushings of patient posterior oropharyngeal walls, as well as ELISA analysis of patient serum with antibodies specific for HPV viral oncoproteins and viral capsid proteins.13
Processed tobacco contains at least 30 known carcinogens. Tobacco smoke contains a high concentration of reactive oxygen species and more than 50 known carcinogens and procarcinogens.14,15 In addition to the direct physical trauma to the upper aerodigestive tract from smoking and drinking, several enzymatic systems are in place to metabolize and deactivate these compounds. It has been proposed that the genetic polymorphisms of these systems to detoxify carcinogens can potentiate putative carcinogens and lead to the development of cancer by either failing to deactivate carcinogens or activating pro-carcinogens.
Cytochrome P-450
The cytochrome P-450 system of enzymes, which is responsible for the oxidative metabolism of most chemicals encountered, consists of more than 20 subfamilies and 60 separate P-450 genes. These enzymes, which are found primarily in the liver tissue, are also found in the aerodigestive tract tissue.16 Of these, the most studied is CYP1A1, which is present in oral tissue.17 Several studies into two specific mutations of this enzyme have revealed only a mild risk association between CYP1A1 exon 7 polymorphism and the development of HNSCC.18 Of particular note, statistical significance was noted only in studies of Asians, whereas results of studies in whites were equivocal. Other systems that have been studied but for which there are insufficient data or inconclusive evidence are CYP1B1, CYP2D6, and CYP2E1.
Glutathione S-Transferase
One of the family of enzymes responsible for the detoxification and processing of such harmful chemicals is known as glutathione S-transferase (GST). There are five separate loci for the production of different GST subtypes that are responsible for detoxifying many different compounds. There also exist several polymorphisms within these subtypes, which may delineate those people more at risk for the development of cancer in the presence of tobacco smoke exposure. In a large meta-analysis, Geisler and Olshan15 reported inconsistent results among 24 studies looking at the role of GSTM1 and GSTT1. They concluded that there are likely subtle effects of polymorphisms on detoxifying enzyme function and the complexity of detoxification processes in general, and that large population-based studies must be performed before the effects of tobacco smoke in subjects with these alterations can be correctly ascertained. In 2003, Hashibe and associates19 performed a pooled analysis and a meta-analysis of studies looking at GSTM1, GSTT1, GSTP1, and CYP1A1. They concluded that in the 33 studies reviewed, there is a modest association of GSTM1 and GSTT1 genotypes with head and neck cancer risk. They further concluded that their pooled analysis “supports the notion of greater risk when multiple GST loci are considered.”19
Other Enzymatic Systems
Variations in other detoxifying enzymes, such as uridine diphosphate–glucuronosyl transferase (UDPGT) have been implicated in an increased risk of head and neck cancer as well.20 A low-activity UDPGT genotype was associated with 3.7-fold greater odds of development of laryngeal cancer in smokers; the odds ratio increased to 6.1 in heavy smokers and was not increased in those who never smoked. Another interesting study, by Elahi and colleagues,21 found that human 8-oxoguanine-DNA glycosylase (hOGG1) polymorphism is an important risk factor for the development of orolaryngeal cancer in smokers and alcohol drinkers. The hOGG1 gene encodes a DNA glycosylase that is involved in the excision repair of 8-hydroxy-2′-deoxyguanine (8-OH-dG) from oxidatively damaged DNA. These researchers found that nonsmoking, nondrinking patients with mutant hOGG1 were not at greater risk of orolaryngeal squamous cell cancer. However, smokers and drinkers with this mutation had 4.8 and 6.9 greater odds of development of orolaryngeal cancer, respectively.21 These two studies suggest that these enzymes are involved in tobacco smoke detoxification and that the genotype can predict the phenotype.
Much study has also been devoted to examining N-acetyltransferases, reduced nicotinamide adenine dinucleotide:quinone oxidoreductase 1, aldehyde dehydrogenases, and alcohol dehydrogenases. Some data implicate these substances in the development of cancer. However, larger studies are required to clearly and definitively determine whether they contribute to the development of cancer.18
In summary, the molecular mechanisms behind tobacco smoke detoxification are beginning to be elucidated, but further work must be performed to enable accurate prediction as to which people are more susceptible to the development of cancer when subjected to this environmental exposure. Gronau and colleagues22 have shown that detoxification enzymes are functionally redundant and that only the simultaneous deficiency of several detoxification enzymes increases the risk for oral cancer. Furthermore, a follow-up study by the same group showed that it is probable that the enzymatic activity rather than the actual genotype can influence the risk of cancer.23 Complicating the issue even more is the influence of ethnicity on the prevalence of polymorphisms; a study in the Netherlands found that two groups of people less than 125 km apart showed statistically significant differences in the prevalence of the GSTT1 genotype.24
Other studies have evaluated the enzymes as prognostic markers. Although these detoxifying enzymes play a role in clearing carcinogens, they may also inactivate chemotherapeutic agents delivered in an attempt to kill offending tumor cells. One study examining a different subtype of GST, GST p, found that those patients with high levels of this enzyme had a worsened overall survival when treated with neoadjuvant chemotherapy, likely owing to the clearance of the drug by the GST p.25 Another study examining GSTT1, a known activator of carcinogens,26 found that individuals with the functional genotype were two times more likely to die from any cause during the course of HNSCC and three times more likely to die from disease-specific causes after adjustment of data for age, stage, and primary therapy.27
Lastly, there has now begun to be some study into the role of saliva and its antioxidative abilities. One group found that the saliva of nonsmoking patients with oral squamous cell carcinoma had highly oxidized DNA and protein, increased levels of reactive nitrogen species, and profoundly reduced levels of antioxidants.28 Other studies have also examined antioxidant levels after tobacco smoke exposure and shown statistically significant decreases in antioxidants, but as yet no definitive link to the development of HNSCC has been demonstrated.29
Human Papillomavirus in Head and Neck Squamous Cell Cancer
Oral HPV infection, like alcohol and tobacco, is now recognized to play an important role in the pathogenesis of a subset of oropharyngeal HNSCCs.30–36 These lesions are primarily from the lingual and palatine tonsils in the oropharynx and, to a lesser extent, the oral cavity.31,34,37–42 Furthermore, HPV-related HNSCC has distinguished itself as a separate entity with better prognosis and response to therapy than those of non–HPV-related HNSCC, which is discussed later. It has also been proposed that oral HPV infection is the reason for the rising incidence of oropharyngeal HNSCC relative to all other anatomic sites in the head and neck. We first review in brief what is known about the molecular biology of HPV and how it relates to cancer.
The human papillomavirus is an approximately 7.9-kilobase, non-enveloped, double-stranded, circular DNA virus. It has a specific tropism for squamous epithelium. The virus infects cells in the basal layer of squamous epithelium, and the different types have been traditionally separated on the basis of tropism for cutaneous and mucosal sites as well as of high, intermediate, and low risk, depending on their association with malignancy.43 The sequences for more than 320 different types of HPV have been identified. Low-risk types, such as HPV-6 and HPV-11, can induce benign hyperproliferations of epithelium such as papillomas and warts. The high-risk types have been implicated in cervical cancer, anogenital cancer, and HNSCC. HPV-16 and HPV-18 are the two types most pertinent to HNSCC and are the focus of this discussion.
The HPV genome typically consists of nine open-reading frame sequences, located on only one of the strands of DNA, and is divided into seven early-phase genes (E) and two late-phase genes (L). The early genes serve to regulate the transcription of DNA, and the late genes encode for proteins involved in viral spread, such as capsid proteins.44 The E1 and E2 gene products are specifically involved in regulating the transcription and replication of viral proteins. The E6 and E7 oncoproteins are normally under control of E2 and E1 inhibitory genes; these inhibitory genes can be deleted or altered upon integration, leading to unchecked transcription of E6 and E7,45 which are then able to disrupt the function of Rb and p53, well-known tumor suppressor genes.46 p53 and Rb are tumor suppressor genes in that they regulate cell-cycle checkpoints at the G1 phase. If these genes are inactivated, cells are more prone to push through division and replication, even in the setting of harmful gene mutations, which can lead to malignancy.
The E6 gene is able to inactivate p5347 through interaction with the E6-associated protein. This complex then binds with p53 and facilitates ubiquitin-dependent degradation of p53.48 E7 is able to bind and interact with the Rb gene product.49 E7 has the ability to phosphorylate the Rb proteins, leading to degradation by ubiquitination.50 This process subsequently leads to E2F activation, which produces a family of transcription factors resulting in cell proliferation. Many other possible mechanisms have been discovered by which these proteins can induce malignancy so that their role in carcinogenesis is ensured.51 It has also been shown that p53 sequence alterations are decreased in the setting of HPV infection, because there is an alternative means of p53 silencing with the production of E6, further delineating HPV-related cancers.31,46,52,53
The idea that HPV causes HNSCC was first proposed when the cytopathic effects of HPV (koilocytosis) were noted on light microscopy of oral lesions.54 In situ hybridization later confirmed the presence of HPV DNA in oral pre-malignancies (4/5 leukoplakias) and malignancies (3/6 carcinomas), thereby suggesting a causal association of HPV and carcinogenesis in oral lesions as well.55 It is now established that histopathologically, HPV-positive tumors tend to have a poorly differentiated and frequently basaloid histology that often lacks keratin.30,31,36,56 Since then, researchers also discovered that human keratinocytes expressing E6 and E7 genes from HPV-16 become immortal,57 as do oral epithelial cells.58–60 It should be noted that the virus must integrate into genomic DNA to have its oncogenic effect.61,62
Further study finally confirmed the link between HPV and oropharyngeal HNSCC. In a 2005 meta-analysis, HPV genomic DNA was detected in approximately 26% of all HNSCCs by sensitive polymerase chain reaction–based methods.63 However, in the majority of studies, 50% or more of oropharyngeal tumors contained the HPV genome.62 In a multinational study conducted by the International Agency for Research on Cancer (IARC), only 18% of oropharyngeal tumors were HPV positive, indicating that this proportion likely varies by geography.64 Regardless of the study population, high-risk HPV-16 accounts for the overwhelming majority (90%-95%) of HPV-positive tumors.63 In addition to the ability to reliably detect HPV in a subset of oropharyngeal cancers, detection of HPV in cervical lymph nodes of patients presenting with an occult primary may be used to establish, with high specificity, the location of the primary within the oropharynx.65 Tonsillectomy has been shown in retrospective analyses to identify the primary site of cervical metastases as the contralateral or ipsilateral tonsil in approximately 10% or 30% of cases, respectively.66–69 Therefore, HPV-related cancer is a distinct established entity that can be reliably diagnosed.
The patient population that presents with HPV-positive HNSCC is also distinct. On average, patients with HPV-positive HNSCC are approximately 5 years younger than patients with HPV-negative HNSCC, with equal distribution between the sexes.41,70–73 HPV-positive HNSCC is more likely than HPV-negative HNSCC to occur in the nonsmoker and nondrinker.31,32,39,40 In a study restricted to patients with oropharyngeal cancers, nonsmokers were approximately 15-fold more likely than smokers to have a diagnosis of HPV-positive HNSCC.74 However, whether there is a synergistic, additive, or no relationship between smoking or alcohol use and HPV infection remains to be seen, and research has supported all three possibilities.
Risk factors for HPV-related HNSCC include a high lifetime number of vaginal-sex partners (26 or more), a high lifetime number of oral-sex partners (6 or more),13 and seropositivity for HPV-16 viral capsid protein antibodies,33 which carries a 15-fold higher risk for HNSCC.
In the majority of studies, patients with HPV-positive tumors have a better prognosis—as much as 60% to 80% lower risk of dying from the cancer—than patients with HPV-negative tumors, after data are controlled for other risk factors.31,71,74–77 The reason for their improved survival is unclear; however, better radiation responsiveness, immune surveillance against viral antigens, and the absence of field cancerization in these patients, who tend to be nonsmokers, have been postulated as possibilities. In addition, E6-related degradation of p53 in HPV-positive cancers may not be functionally equivalent to HPV-negative p53 mutations, and therefore, HPV-positive tumors may have an intact apoptotic response to radiation and chemotherapy.
Therapeutic implications of an HPV-positive diagnosis are an active area of investigation. The belief that HPV-positive HNSCC is more responsive to treatment than HPV-negative HNSCC is being considered in the design of future clinical trials. A clinical trial of an HPV-16–specific therapeutic vaccine is also currently being evaluated. The vaccine is administered in the adjuvant setting and is intended to enhance the cytotoxic T-cell response to the HPV-16 oncoproteins.78 With regard to prevention, a prophylactic vaccine composed of the HPV-16 viral capsid protein has been shown to prevent persistent HPV-16 infection and the development of cervical dysplasia.79,80 However, the clinical trials have not included an evaluation of the impact of the vaccine on oral HPV infection. The vaccine does have the potential to affect HNSCC incidence, because the current vaccines are targeted to HPV-16 and because animal models demonstrate the vaccine to have a protective effect and a reduction in the development of HPV-related oral lesions.
Possible future diagnostic tests that would likely have high specificity but low sensitivity for a diagnosis of HPV-associated HNSCC include the detection of HPV-16 DNA in plasma,81 which can be used for surveillance. Other screening tests, such as fluorescence in situ hybridization (FISH) on Papanicolaou smears obtained directly from tumors and HPV-16 E6 and E7 seroreactivity, are currently being tested. And the impact of a positive test result on a patient’s risk for the development of cancer is still unclear.
Chemoprevention
As discussed previously, there are well-defined risk factors for the development of HNSCC. In addition to avoiding exposures, there is the possibility of both preventing the development of HNSCC and preventing progression of early disease. Currently, most of the therapy for head and neck cancer is focused on treating the disease after it has already progressed to malignancy. However, because head and neck cancer often manifests initially as premalignant disease and because there is a defined set of high-risk individuals, many researchers have begun the search for agents that can prevent disease from occurring, prevent recurrent disease, or prevent disease progression.82
The concept of chemoprevention was first proposed by Sporn and colleagues,83 who looked at the protective effects of vitamin A and its derivatives on carcinogenesis. The mechanism of action was thought to be via binding of retinoic acid receptor molecules,84,85 which then function as angiogenic and growth-regulating factors.86,87 In a landmark study, the utilization of high-dose 13-cis retinoic acid in patients with known head and neck cancers proved helpful in preventing the development of second primary tumors88 and in the treatment of oral leukoplakias,89 although significant toxicity was associated with this therapy, including skin lesions, conjunctivitis, and cheilitis. Furthermore, when therapy was discontinued, the risk of disease development returned to baseline after about 2 years.90 A follow-up study looked at use of low-dose fenretinide (a synthetic retinoid) for leukoplakia, and early results were promising for prevention of malignant transformation.91 Unfortunately, the trial was ended because of low recruitment. In a large trial, 1384 patients were treated with isotretinoin to prevent second primary HNSCC. Treatment showed no benefit, and only smoking cessation correlated with a decreased risk of recurrence.92 Another large trial by the Italian Head and Neck Chemoprevention Study Group found that treatment with 13-cis retinoic acid for 1 year after resection showed no benefit.93 However, the question is not completely answered; there has been evidence that in cell lines, the combination of 13-cis retinoic acid, interferon-alpha-2a, and alpha-tocopherol can cause cell cycle arrest and apoptosis.94 Another study showed that a synthetic retinoid, MX3350-1, could induce the extrinsic and intrinsic pathways in head and neck squamous carcinoma cells.95 Whether this combination or any other combination will be beneficial still remains to be seen.
Cyclooxygenase-1 (COX-1) and COX-2 inhibitors have also been studied as possible mediators of prostaglandins and the inflammation that often accompanies many different tumors. Prostaglandin levels have long been known to be raised in head and neck cancers as well, presumably owing to the up-regulation of COX-1 and COX-2.96,97 Animal models suggested that aspirin or ibuprofen could inhibit the formation of buccal tumors.98,99 Colon cancer was among the first tumors studied widely in humans, beginning with epidemiologic evidence that aspirin reduced the risk of colon cancer development.100 Sulindac was shown prospectively to reduce the number and size of colonic polyps in patients afflicted with this disease.101 Studies of the use of these agents in colorectal, breast, lung, and esophageal carcinomas followed. The mechanism behind the effect of COX-2 inhibitors appears to be multifactorial.102,103 The overexpression of COX-2 leads to decreased apoptosis, thereby prolonging the life span of abnormal cells. Furthermore, inflammation may be a risk factor for epithelial carcinogenesis because it is often found in these malignancies. Angiogenesis may also be tied into the COX-2 pathway, because vascular endothelial growth factor (VEGF) expression is elevated by high COX-2 levels.104 Thus, there are several mechanistic reasons why COX-2 inhibitors would be beneficial in preventing recurrence, development, or progression of head and neck lesions, especially in high-risk patients. There have been some promising results in animal models, but study in humans has only begun.105 In addition, the use of epidermal growth factor receptor (EGFR) inhibitors as chemopreventive agents, both individually and in combination, is also being studied.105
Genetic Alterations
Cytogenetics
The most basic form of cytogenetics is karyotype analysis, which entails a visual analysis of cells that have undergone short-term culture and have been arrested in the metaphase state to screen for abnormalities in copy number gain or loss and chromosomal breaks or rearrangements. Data have confirmed frequent loss of material on chromosomes 3p, 8p, 11q, 13p, 14p, and 15p with gains on chromosomes 1q, 3q, 8q, and 15q.106 The limitation of karyotype analysis lies in its resolution. Each region carries numerous genes that could be lost or gained.
Other techniques with different strengths and weaknesses have been employed to evaluate the tumor genome. Spectral karyotyping (SKY) adds the use of chromophores to karyotype analysis; it provides a unique fluorescent background for each of the chromosomes. SKY makes characterizing complex rearrangements or chromosomal aberrations easier. The technique of SKY has been used on HNSCC, and frequent losses were identified at 3p, 8p, 9p, 11q, 14p, and 15p along with gains at 1q, 3q, 8q, and 15q.107 Comparative genomic hybridization (CGH) is a technique utilized to detect unbalanced chromosomal gains or losses whereby normal DNA and tumor DNA are labeled and hybridized to normal metaphase chromosomes. The relative fluorescence at the different regions is determined, and regions of loss or gain can be detected. The resolution is accurate to about 20 megabases. FISH uses sequence-specific fluorescent probes that are detected through light microscopy after hybridization. It also allows for the specific detection of areas of loss and gain; however, FISH is limited in its genome-wide application.
The use of a combination of SKY, FISH, and CGH on the same samples has the benefit of balancing out the weaknesses and ensuring the consistency of these techniques. Several investigators have used such an approach in evaluating HNSCC.108,109 As in other studies, common chromosomal losses were found to occur at 1p, 3p, 4p, 8p, 10p, and 18q with gains at 3q, 5p, 8q, 9q, and 14q, and amplification at 3q13, 3q25-26, 5q22-23, 7q21, 8q24, 11q13-14, 12p13, 14q24, and 20q13.1. There was good correlation between SKY and CGH results, and FISH was generally employed to further delineate the nature of aberrations with its sequence specificity. One study identified PIK3CA, a member of the Akt pathway, as a candidate oncogene on chromosome 3q involved in HNSCC.109
Cytogenetic alterations may also predict prognosis. CGH analysis of 82 primary head and neck carcinoma samples demonstrated that amplification of 11q13 correlated with a poor outcome.110 A similar finding was shown in 12 laryngeal cancer cell lines whereby 11q13 amplification was associated with shorter patient survival.111 Thus, these alterations can be used as markers for prognosis, because they are associated with genetic changes that can influence the development or aggressiveness of HNSCC.
With the advent of microarray technology, the application of cytogenetics has begun to take on a new form. CGH-array now hybridizes the normal DNA and tumor DNA to millions of probe sequences, allowing for a much finer map down to 5 to 1010 kilobases. This technique has been shown to have application in HNSCC112,113 and has been used to identify a novel region of gain on 8q22 and the LRP12 gene.114
Single Nucleotide Polymorphisms
Single nucleotide polymorphisms (SNPs) are DNA sequence variations that occur when one nucleotide in the genome sequence is altered. For a variation to be considered an SNP, it must occur in at least 1% of the population. SNPs are scattered throughout the coding and noncoding regions of the genome and account for about 90% of all human genetic variation. Although most SNPs have no effect on cell function, these variations in the DNA sequence can have several uses. Because SNPs are highly conserved, they can be used for profiling cancers and subsequently as clinical biomarkers. In addition, SNPs have been used to reliably map out the locations of genes of interest. For instance, SNP arrays have been created to scan the entire genome, have been used to provide finer mapping of areas of loss or gain, and have identified a region on 6q that is implicated in some cases of HNSCC.115
Microsatellites
Microsatellite repeats are tandem repeats of noncoding sequences scattered throughout the genome. Use of these conserved regions as markers in the human genome enabled areas of loss to be found in cancer. Once a microsatellite was found to be deleted or missing in cancer in comparison with normal tissue, the region was identified as having a possible TSG in its proximity. Finer mapping or sequencing could then be performed to identify the gene of interest. Out of this technique came the HNSCC progression model that was discussed in the chapter’s introduction. This study by Califano and colleagues5 demonstrated that TSG deletions occurred early in the precancerous lesions and that there was an accumulation of alterations with progressive disease in HNSCC.
Oncogenes
Matrix metalloproteinases (MMPs) are genes encoding for a variety of proteins that are thought to be involved in cell adhesion, proliferation, and migration. MMP-2 was one of the first in the family noted to be related to lymph node metastasis in HNSCC,116 but several other studies have seen other members of MMP overexpression.35,117 MMP expression may also be predictive of advanced disease and tumor metastasis in HNSCC.35,117–119 Interestingly, MMP expression may be influenced by EGFR up-regulation,120 so there is likely to be overlap between many of the pathways involved in HNSCC. Furthermore, it appears that there are stromal elements that produce MMPs and thus could play a role in the development of metastatic disease.121
One of the p53 family members, ΔNp63-α, is known to be amplified and overexpressed in HNSCC cell lines and to have oncogenic properties.122 It is also known to be created through the alternative splicing of the p63 protein, which encodes for several other putative oncogenes. One possible mechanism of action of ΔNp63-α is through the accumulation of the transcription factor β-catenin.123 Later data demonstrate that ΔNp63-α functions through the inhibition of p73, and it is through this inhibition that ΔNp63-α is able to repress apoptosis via bcl-2.124
Tumor Suppressor Genes
One of the earliest significant TSGs identified in HNSCC was p53. A transcription factor the gene for which has been mapped to chromosome 17p13, p53 is activated in times of cellular stress such as irradiation, hypoxia, and environmental toxin exposure. Interestingly, early reporters of p53 believed that it was an oncogene, because it was found to be overexpressed in cancers. However, it is now known that the p53 gene is inactivated most often by point mutations, although overexpression of the mutant forms still occurs.125 Activated p53 serves to send cells into apoptotic pathways or can regulate growth through cell cycle control; thus, it plays a critical role in the maintenance and proliferation of cancer cells. Indeed, p53 is the most commonly mutated TSG among all human cancers,126 and HNSCC is no exception.127 Premalignant head and neck lesions have also been shown to harbor p53 mutations, suggesting that this mutation can be an early event.128 Mutations in p53 have been correlated with poorer survival and poorer response to treatment.129–133 Thus, p53 serves as an excellent marker for disease and has been the target of many therapeutic interventions.
As described previously, cytogenetic data indicated that deletion of 9p21 is a common event in HNSCC, and the LOH data confirmed that it is a common, early event.134,135 The CDKN2a locus codes for p16 and p14(ARF), which have been accepted as the relevant genes contributing to HNSCC. The p16 gene plays a role in cell cycle regulation at the G1 state through cyclin-dependent kinases 4 and 6, and the p14(ARF) gene is thought to function in the stabilization of p53 through MDM2, a gene that is a negative regulator of p53. Thus, there are two prominent TSGs at this locus that show mechanistic evidence of their function in regulating cell growth and division. Studies suggest that loss of p16 expression may be associated with poorer prognosis in patients with HNSCC.136,137 Although p16 was frequently found to be down-regulated in HNSCC, there was a surprisingly low incidence of point mutations or homozygous deletions that would explain its contribution to HNSCC.138 The tumor-suppressive function of p16 was indeed confirmed, although it became evident that there were other mechanisms involved in its silencing (discussed later).
The deleted in colorectal cancer (DCC) gene is located on chromosome 18q21, a region that was shown to have LOH in dysplastic head and neck lesions,139 in oral cancer cell lines,140 and in a significant percentage of primary HNSCC.141 Furthermore, loss of this region was shown to be more common in the metastatic and locally recurrent lesions, suggesting that it played a significant role in more aggressive lesions.142
However, there did not appear to be a higher incidence of tumor formation in heterozygous DCC mice than in homozygous deleted DCC mice. Thus, there was some confusion as to its true role in tumorigenesis. One study demonstrated that DCC’s function was related to binding with netrin-1 and thus that DCC acts as a conditional TSG. Gene silencing via promoter hypermethylation was confirmed in primary HNSCC and cell lines, and subsequent growth assays showed that transfection of DCC into DCC-cell lines was able to arrest growth, whereas the addition of netrin-1 led to the resumption of cell growth.143 Thus, the mechanism of the tumor-suppressive effects of DCC is not as simple as that of other models and also suggests that there may be many other ways in which TSGs may function.
Tumor Suppressor Gene in Fanconi’s Anemia
Fanconi’s anemia is an autosomal recessive DNA repair disorder that leaves patients uniquely vulnerable to DNA damage and the subsequent development of a multitude of cancers. Thus far, 11 genes have been identified in this disorder that ultimately are involved in the regulation of the BRCA-1 and BRCA-2 pathway (involved in the repair of double-stranded DNA breaks). Previously, patients succumbed to aplastic crises or hematologic malignancies. However, with advances in bone marrow transplantation and longer life spans, patients with Fanconi’s anemia have been found to be at profound risk for development of various cancers, in particular HNSCCs. Patients with this disorder are atypically young (<50 years of age) and are usually women who do not smoke tobacco or drink alcohol. The relative risk of HNSCC in patients with Fanconi’s anemia is 700 times greater than that in the general population.144 After bone marrow transplantation, which is performed to prevent a fatal aplastic crisis or hematologic malignancy, the risk is further increased by fourfold.145 Patients with this disorder should be treated with surgery rather than irradiation or chemotherapy, to prevent further DNA damage and the development of other malignancies.
Epigenetics
The field of epigenetics has greatly furthered our understanding of not only cancer but all of molecular biology. Epigenetics is a term used to describe the regulation of gene expression that does not involve DNA sequence alterations but, rather, is a heritable modification of the DNA molecule itself and/or the proteins associated with it. One way in which this modification can occur is through the covalent addition of a methyl group to the 5′ carbon of the cytosine ring, resulting in 5-methylcytosine. However, in order for this methyl group to be added, a cytosine base pair must be followed by a guanine base pair in the DNA sequence, a so-called CpG dinucleotide. Overall, CpG dinucleotides occur at lower than expected frequencies throughout the human genome. However, they are found more frequently in the promoter regions of genes as small stretches of DNA called CpG islands.146 The addition of the methyl group to the cytosine prevents binding of transcriptional activators and RNA polymerase, effectively silencing the gene. Once methylated, the modification is then passed to progenitor cells, making it a heritable or permanent modification to gene expression. DNA methyltransferases (DNMTs) are responsible for maintaining the methylation status of a gene in the progeny. Because CpG islands are most commonly found in promoter regions of the genome, they are believed to be one mechanism of gene expression regulation. In summary, hypermethylation and hypomethylation are epigenetic means of regulating gene expression (Fig. 76-1).
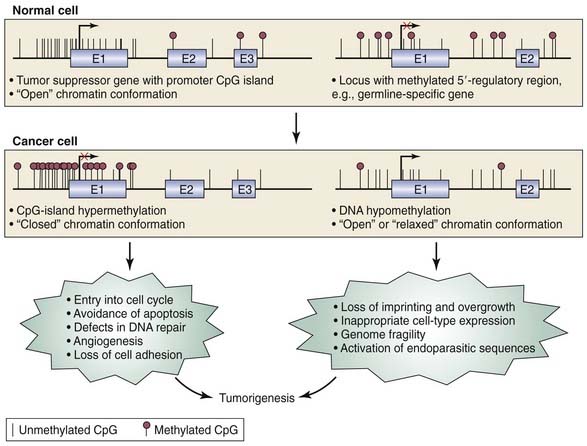
In addition to the methylation of promoters, histones have been shown to play an active role in regulating gene expression. Once thought to be static packaging proteins, histones are now recognized to be dynamic molecules that can physically determine whether transcription factors have access to promoters. Histones may undergo many post-translational chemical modifications, such as acetylation, methylation, phosphorylation, ubiquitylation, and sumoylation.147 Histone modifications and DNA methylation play a significant role in the organization of nuclear structure, ultimately influencing what genes may be suppressed or expressed.148
The phenomenon of methylation as a regulator of gene expression was first recognized in development, during which the coordinated expression and silencing of genes needs to take place in an organized fashion.149 It was quickly proposed that epigenetic control of TSGs, as a novel mechanism of gene regulation, could be an important mechanism of carcinogenesis.150,151 Initially, studies of the global methylation status were performed in animals, and cell culture showed mixed results. Many workers believed that the observed hypomethylation of tumors was simply a marker of an actively dividing cell or the result of malignant transformation rather than the cause. But Feinberg and Vogelstein150,151 were the first to analyze primary tissue from resected cancers and examine both global methylation and methylation of specific known oncogenes. Interestingly, they found that cancers were globally hypomethylated (10%) and that Ras oncogenes were specifically hypomethylated.
Although hypomethylation was the originally identified epigenetic change in cancer, it was overlooked in preference for hypermethylation for many years and is only now undergoing a renaissance. This situation occurred, in part, because of previous bias in experimental design; if one looks for altered methylation only at sites that are normally unmethylated, then one will observe only hypermethylation.152 Furthermore, the early success of Baylin and Herman (discussed later) in finding specific regions of hypermethylation prompted many other researchers to search for similar regions with similar techniques.
One of the earliest genes to be characterized as being under epigenetic control is the retinoblastoma gene. Looking in primary tissues of resected retinoblastomas, Sakai and coworkers153 found that in 10% of the tumors analyzed, the promoter of Rb was hypermethylated in the absence of any other mutations. Since that discovery, many different tumor types have been shown to exhibit various TSG methylation.
Significant promoter hypermethylation has been demonstrated in many of the known TSGs in head and neck cancer. One of the more interesting findings is the high incidence of p16 hypermethylation. It has long been puzzling that p16 expression has been found to be decreased in a majority of HNSCCs, whereas point mutations and homozygous deletion are rare and the incidence of LOH is not. Promoter hypermethylation of p16 has been shown to be quite frequent in HNSCC, explaining the apparent discrepancy between the incidence of deletions or mutations and low expression.146,153–156 Thus, Knudson’s “two-hit” hypothesis could include promoter hypermethylation as one of the hits along with the more traditional sequence mutation or chromosomal deletion.
Many other genes have emerged as putative TSGs in HNSCC based on promoter hypermethylation, including lhx-6 (DIME-6), ATM, p15, TIMP-3, MGMT, RARB-2, DAP-K, E-cadherin, Cyclin A1, RASSF1A, and DCC.157–167 The list of TSGs that are silenced through promoter hypermethylation continues to grow every year. Furthermore, it has been recognized that many other avenues of research cross paths with epigenetics. For instance, microRNAs (small non-coding RNA molecules; see later) that have tumor-suppressor function have been demonstrated to also undergo DNA methylation–associated silencing in cancer and therefore are under the same epigenetic control164,168,169 (see Fig. 76-1).
However, one must recall that in general, DNA is hypomethylated in cancer despite local regions of hypermethylation. This apparent paradox was attributed to overall cell activation after carcinogenesis was initiated. Evidence is now emerging on the importance of the hypomethylation of genes. Studies have now shown that the regions of hypomethylation correlate with genes that were important to development and were subsequently “turned off” by methylation. These genes, once reactivated, have proved to be oncogenic. Some of these genes are HOX11, MAGE family, CAGE-1, uPA, MTS1/S100A4, MASPIN/SERPINB5, and BCSG/SNCG.170 Specific data on genes that are activated in HNSCC are yet to be published, although a 2007 study demonstrated that there is indeed global hypomethylation in HNSCC.171
The natural question regarding any of the previously mentioned discoveries regarding epigenetics is how they will ultimately affect clinical practice, if at all. Translational epigenomics has been an exciting area of research that has grown with the field of epigenetics itself. There are several methods to translate epigenetic discoveries into clinical practice. The first is the use of the epigenetic profile, which refers to the fact that unique profiles of hypermethylation define each cancer type.172,173 Once defined, the profile of HNSCC could be used as a marker of prognosis and possibly to predict responsiveness to treatment. Second, the hypermethylated genes themselves are potential therapeutic targets. Promoter hypermethylation is an attractive route for therapeutic intervention because it does not involve loss of DNA or sequence alterations. Selective demethylation is not feasible at this point, but it could eventually become a reasonable means of cancer therapy. Third, detection of methylated DNA in body fluids is a potential marker for screening and surveillance. Studies have already proven the ability of such markers to be detected, but further work is needed to improve their sensitivity and specificity.165,174 Lastly, it may be possible, with the development of more rapid detection strategies, to perform molecular margin analysis of cancers. These topics are more thoroughly addressed in the molecular diagnostics section of this chapter.
Epidermal Growth Factor Receptor
One of the best-studied areas of the molecular biology of HNSCC is the role of EGFR and its associated pathways in carcinogenesis. Expression of EGFR is a normal finding in many tissues, including the dermis, gastrointestinal tract, and kidneys. However, dysfunction of this receptor and its associated pathways occurs in most epithelial cancers175 and in 80% to 90% of HNSCCs specifically.176,177 What is unique about this area of inquiry is that the research into the biology of EGFR has led directly to clinically beneficial targeted therapies. Before delving into therapies, we examine the basic molecular biology of EGFR.
Biology
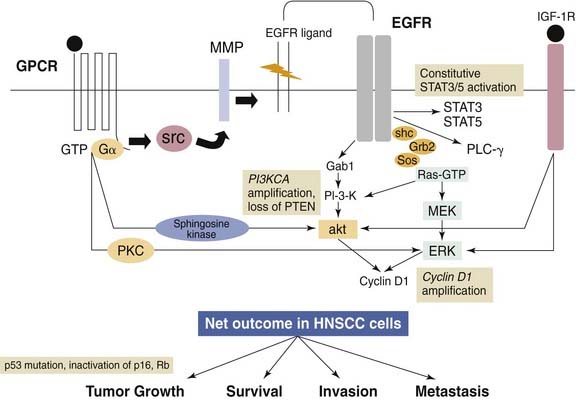
A general understanding of the EGFR-related pathways is necessary to comprehension of the therapies directed against it (Fig. 76-2). EGFR is a cell surface receptor that is a member of the larger ErbB family of proteins, which includes the Her2, Her3, and Her4 receptors. EGFR possesses an extracellular ligand-binding domain, a transmembrane region, and an intracellular domain. The intracellular domain includes the tyrosine kinase domain and multiple phosphorylation sites. Once the extracellular portion of the receptor binds a ligand, homodimerization or heterodimerization of EGFR with other ErbBs follows, which then results in the autophosphorylation of the receptor. Currently, 12 ligands of the ErbB family have been identified. Once phosphorylated, the receptor can signal via the MAPK, Akt, ERK, and Jak/STAT pathways. These pathways are related to cellular proliferation, apoptosis, invasion, angiogenesis, and metastasis.175,177,178 There is also some evidence that the receptor may localize to the nucleus and act as a transcriptional activator. Finally, one study has even linked the effects of tobacco smoke to increased EGFR expression and cellular proliferation.
Clinical Implications
The pathologic manifestations of EGFR in HNSCC are achieved by at least four major mechanisms, as follows: (1) overexpression of EGFR ligands and establishment of autocrine/paracrine loops, (2) mutational activation of EGFR, (3) amplification of EGFR, and (4) transactivation by other receptor and nonreceptor tyrosine kinases. Although EGFR-driven molecular pathway aberrations are important and are altered in 90% of HNSCCs, they cannot be considered the dominant or sole genetic alteration in the molecular pathogenesis of HNSCC. Initially, EGFR was first found to be up-regulated in HNSCC cell lines179 as well as in a high percentage of primary HNSCCs.179–181
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


