CHAPTER 49 Benign Tumors of the Sinonasal Tract
Significant refinements in imaging techniques and the widespread application of endoscopic surgery have led to revived interest in the management of benign tumors of the sinonasal tract. Not unexpectedly, this anatomic region may be involved by a large variety of different histopathologic entities that, according to the World Health Organization (WHO) classification, include epithelial tumors (papillomas and salivary gland–type adenomas), soft tissue tumors (myxoma, leiomyoma, hemangioma, Schwannoma, neurofibroma, meningioma), and tumors of bone and cartilage (giant cell lesion, giant cell tumor, chondroma, osteoma, chondroblastoma, chondromyxoid fibroma, osteochondroma, osteoid osteoma, osteoblastoma, ameloblastoma, nasal chondromesenchymal hamartoma).1 Although originating from the pterygomaxillary fossa, juvenile angiofibroma is habitually included in the group of sinonasal tract tumors because of its common presentation as a nasal or nasopharyngeal mass.
This chapter discusses in detail inverted papilloma, osteoma, and juvenile angiofibroma, which are the most common histologic types seen in our series of 635 benign tumors of the sinonasal tract encountered over 15 years at two University Hospitals (Table 49-1). We also provide information on other benign tumors of less common observation.
Inverted Papilloma
Inverted papilloma (or Schneiderian papilloma, inverted type), which is included in the group of sinonasal papillomas together with the oncocytic and exophytic variants, is the second most common benign tumor of the sinonasal tract after osteoma, even though it represents the most common surgical indication for a benign sinonasal tumor. The lesion is estimated to represent 0.4% to 4.7% of all surgically removed nasal tumors, with an incidence ranging from 0.6 to 1.5 cases per 100,000 inhabitants per year.2,3 Men are more commonly affected than women, and the lesion is prevalently observed in the fifth and sixth decades of life.
With regard to the histologic appearance, inverted papilloma is composed exclusively or almost exclusively of hyperplastic ribbons of basement membrane–enclosed epithelium that grow endophytically into the underlying stroma. The epithelium is multilayered and formed of squamous or ciliated columnar cells mixed with mucocytes.4
The association of inverted papilloma with squamous cell carcinoma has been overemphasized, with reported frequencies as high as 56%.5 Recent data clearly show that the prevalence varies between 3.4%6 and 9.7%7 and that synchronous occurrence is more common than metachronous.
Inverted papilloma is suspected to have a viral etiology. Human papillomavirus DNA has been demonstrated by in situ hybridization or the polymerase chain reaction in papillomas, with a prevalence of serotypes 6, 11, 16, and 18.8,9 The last two serotypes have been specifically found to be associated with inverted papillomas that show histologic signs of malignant transformation. Not unexpectedly, in this setting an increase in levels of epidermal growth factor receptor and tumor growth factor-α has been demonstrated.10
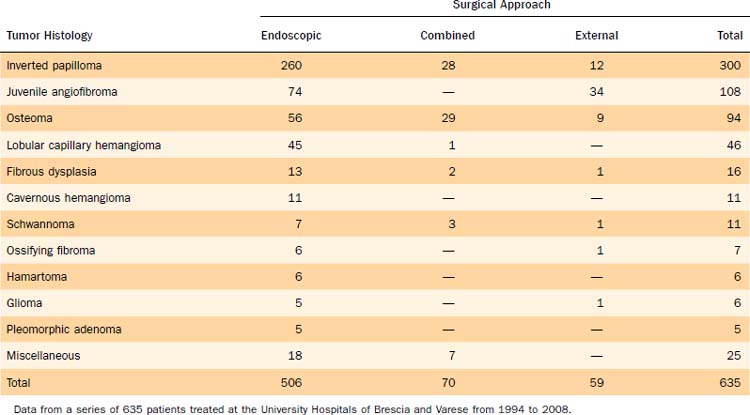
Unilateral nasal obstruction with watery rhinorrhea is the most common symptom prompting the patient to seek otolaryngologic consultation, whereas epiphora, proptosis, diplopia, and headache may be associated with advanced lesions involving the orbit or the skull base. Endoscopy of the nose, usually showing a pale, polypoid lesion with a papillary appearance protruding from the middle meatus (Fig. 49-1), may easily suggest the diagnosis, which is sometimes made less obvious by the concomitant presence of inflammatory polyps. A biopsy performed under endoscopic guidance is indicated to establish the histologic diagnosis.
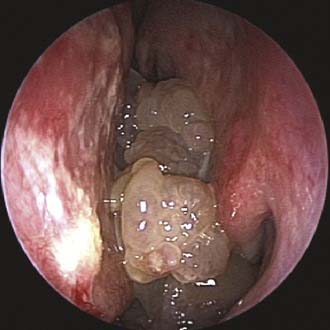
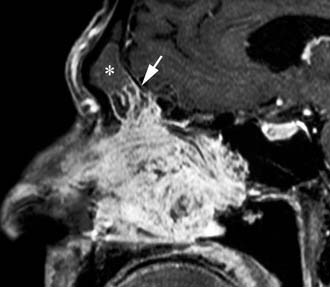
Imaging studies are required to assess the extent and three-dimensional configuration of the lesion and to disclose its relationship with surrounding structures (i.e., orbit, skull base, optic nerve, internal carotid artery). In our experience, these goals are best achieved by MRI with gadolinium enhancement, which also has the advantage over CT of better differentiating tumor from inflammatory mucosal changes and to disclose the so-called cerebriform-columnar pattern (Fig. 49-2). This reflects the histologic arrangement of inverted papilloma characterized by the alternation of regular parallel folds made of a highly cellular metaplastic epithelium and of an underlying less cellular stroma. It is therefore highly predictive for inverted papilloma diagnosis.11 However, even MRI has limitations, especially in lesions completely filling the maxillary, sphenoid, or frontal sinus, in differentiating inverted papillomas growing inside the sinus but arising from a small area of insertion from those extensively involving the mucosa. Recent studies indicate that focal hyperostosis12 and osteitic changes13 upon CT may be considered reliable predictors of tumor origin. These findings may also be identified by MRI (Fig. 49-3).
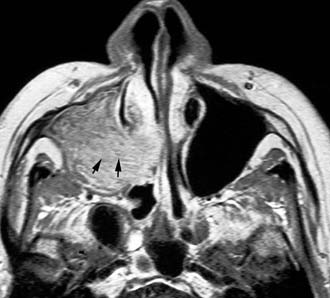
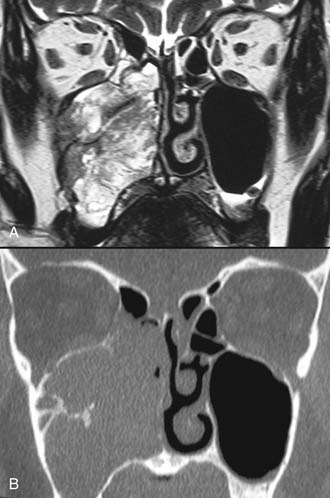
As clearly shown by a 2006 meta-analysis14 and by our experience based on 300 inverted papillomas, endoscopic surgery is a reliable alternative to traditional external techniques for the vast majority of lesions. An exclusive endoscopic approach may be contraindicated in the following situations: (1) massive involvement of the mucosa of the frontal sinus and/or of a supraorbital cell, (2) intradural extension or transorbital extension, both of which are very uncommon situations usually found in patients who have already undergone one or more surgical procedures, (3) concomitant presence of a malignancy involving critical areas, and (4) presence of abundant scar tissue from previous surgery.
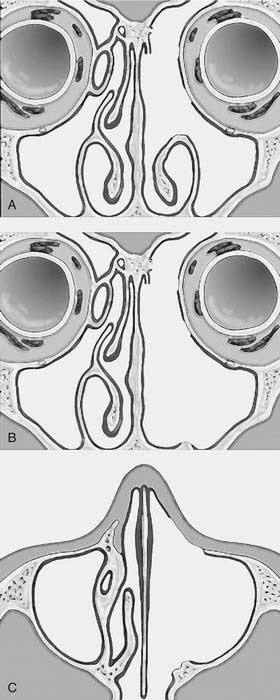
One criticism of endoscopic approaches is the impossibility in always obtaining an en bloc resection. However, it is not the concept of en bloc resection per se that has to be fulfilled to achieve complete removal; the key point is to dissect the involved mucosa along the subperiosteal plane and to drill the underlying bone whenever required by imaging and/or intraoperative findings.15 The extent of the operation is dictated by the site of the lesion and the area of mucosa involved by the lesion. Apart from cases with a clearly identifiable small attachment of the lesion, which can be managed with a very conservative approach,16 there are basically three different types of endoscopic resections available, according to our classification.17 Type I resection (Fig. 49-4A) is indicated for inverted papillomas involving the middle meatus, ethmoid, superior meatus, sphenoid sinus, or a combination of these structures; even lesions protruding into the maxillary sinus without direct involvement of the mucosa are amenable to this approach. Type II resection (Fig. 49-4B), which corresponds to an endoscopic medial maxillectomy, is indicated for tumors originating within the nasoethmoidal complex and secondarily extending into the maxillary sinus or for primary maxillary lesions not involving the anterior and lateral walls of the sinus itself. The nasolacrimal duct can be included in the specimen to increase the exposure of the anterior part of the maxillary sinus. Type III resection (Fig. 49-4C), also known as the Sturman-Canfield operation or endonasal Denker operation,18 entails removal of the medial portion of the anterior wall of the maxillary sinus to enable access to all the antrum walls. It is therefore recommended for inverted papillomas extensively involving the anterior compartment of the maxillary sinus.
Frontal sinus involvement can be observed in different scenarios, ranging from a limited lesion marginally growing from the ethmoid into the frontal recess (Fig. 49-5) to very complex cases in which most or all of the sinus mucosa is diseased. Because the key point for ultimate success is removal of the lesion along the subperiosteal plane with the possibility to drill out the underlying bone, the surgical choice may involve a Draf IIB or Draf III endoscopic sinusotomy or a combination of an endoscopic approach with external sinusotomy through an osteoplastic flap depending on the extent of frontal sinus mucosa involvement, a finding that can usually be definitively assessed only at the time of surgery. Even the involvement of an extensively pneumatized supraorbital cell is frequently challenging. Exposure through the nose may be increased by coagulation and sectioning of the anterior ethmoid artery and by displacement of the orbit after drilling of the upper portion of the lamina papyracea. However, whenever the lesion involves a cell that extends far posteriorly and/or laterally over the orbit and complete resection cannot be achieved transnasally, the surgeon should resort to a frontal osteoplastic flap.
In the era when transnasal resection without endoscopic or microscopic assistance was the most commonly used technique, the rate of recurrences ranged from 40%19 to 78%.20 These extremely high values indicate that these “recurrences,” mostly occurring at the site of primary resection, should have been more appropriately regarded as residual lesions. They reflected the inadequacy of transnasal surgery in affording a radical excision of the lesion. In view of such limitations, medial maxillectomy through lateral rhinotomy was established in the 1970s and 1980s as the gold standard for treatment of inverted papilloma. Although the frequency of recurrences decreased, reportedly from 0%21 to 29%,22 this technique was associated with potential aesthetic sequelae. Therefore, midfacial degloving became the most popular approach for the management of inverted papilloma. The routine use of available staging systems specific for inverted papilloma23–27 would facilitate the comparison of results among different institutions.
Juvenile Angiofibroma
Juvenile angiofibroma is a benign lesion histologically characterized by vascular endothelium–lined spaces embedded in a fibrous stroma that typically affects young male adolescents. Immunohistochemical and electron microscopy studies suggested that the lesion could be considered a vascular malformation (or hamartoma) rather than a tumor.28 These observations led Schick and colleagues29 to postulate that juvenile angiofibroma might develop from incomplete regression of a branchial artery, which arises in embryogenesis between day 22 and 24 and forms a temporary connection between the ventral aorta and dorsal aorta. This artery commonly regresses and forms a vascular plexus that either involutes or may leave remnants, potentially leading to development of juvenile angiofibroma (Fig. 49-6). This theory is supported by the finding that juvenile angiofibroma vessels express laminin α2, which is considered a marker for early angiogenesis.30
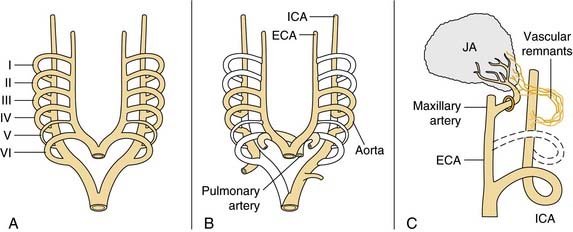
Figure 49-6. Schematic drawing summarizing the theory postulated by Schick and colleagues29 on the origin of juvenile angiofibroma (JA) from remnants of the branchial artery. A, During embryogenesis the sixth branchial arch arteries temporarily connect the ventral and dorsal aortas. B, Physiologic regression of several vascular structures (white vessels) subsequently occurs, leading to definitive configuration of the vascular system. C, Regression of the first branchial arch artery takes place via the formation of a vascular plexus, which is usually complete at birth. Incomplete regression of this structure can explain the typical blood supply for juvenile angiofibromas, from the maxillary and sphenopalatine arteries, with persistent vascular connections to the internal carotid pathway. I through VI, six branchial arteries; ECA, external carotid artery; ICA, internal carotid artery.
The lesion has a pathognomonic epicenter of origin at the level of the pterygopalatine fossa and subsequently grows through different pathways of spread that typically follow foramina and fissures of the skull base. The bone may be involved basically with two mechanisms, resorption by pressure coming from subperiosteal growth or invasion of the cancellous component initially at the level of the root of the pterygoid process, with subsequent expansion within the greater wing and erosion of the floor of the middle cranial fossa. In its early phase, juvenile angiofibroma extends through the sphenopalatine foramen into the nasopharynx and the nasal cavity and along the vidian nerve into the floor of the sphenoid sinus. Lateral extension through the pterygomaxillary fissure leads to invasion of the infratemporal fossa, which in advanced lesions may be completely filled. When the lesion expands anteriorly, the posterior wall of the maxillary sinus is progressively pushed forward. Although benign, juvenile angiofibroma may extend intracranially through the orbit via the inferior and superior orbital fissure or along the maxillary nerve to the parasellar region. Encroachment upon the anterior skull base through the ethmoid is less commonly observed. Regardless of the site and pattern of intracranial involvement, transdural growth of the lesion is very rare.31
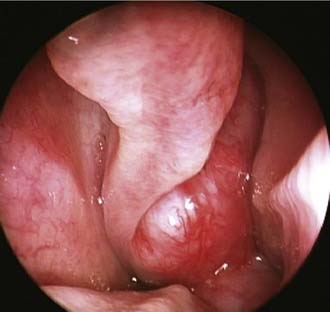
Unilateral nasal obstruction and epistaxis are the most common heralding symptoms of small to intermediate-size juvenile angiofibromas. In advanced lesions, swelling of the cheek, proptosis, or headache may be present, indicating an involvement of the infratemporal fossa, the orbit, or the cranial fossa, respectively. The endoscopic finding of a smooth, hypervascularized lesion originating behind the middle turbinate, which is usually laterally displaced against the lateral wall (Fig. 49-7), in a teenage boy strongly suggests a diagnosis of juvenile angiofibroma, which is usually confirmed by CT and MRI. Therefore, resorting to a biopsy, which is associated with a high risk of hemorrhage, is rarely if ever justified.
On CT and MRI the diagnosis of juvenile angiofibroma is based on the following three features: the area of origin invariably located at the level of pterygopalatine fossa, its hypervascular appearance after contrast enhancement, and its pattern of growth.32 On MRI, the presence on both T1- and T2-weighted sequences of several signal voids within the lesion, indicating major intralesional vessels, further corroborates a diagnosis of juvenile angiofibroma. Sometimes differentiation of this lesion from lobular capillary hemangioma, hemangiopericytoma, and schwannoma can be difficult in view of a similar pattern of enhancement. However, these other lesions do not commonly involve the pterygopalatine fossa and occur in a different age group.
Intraoperative bleeding has always been considered one of the most challenging issues in the management of juvenile angiofibroma, leading in the past to a high rate of persistent disease and to a notable morbidity. The introduction in the early 1970s of preoperative embolization,33 which is commonly performed 48 hours before surgery, has revolutionized the treatment of this lesion by dramatically decreasing intraoperative bleeding and therefore making the assessment of tumor borders at dissection more accurate. Nowadays, the availability of intra-arterial digital subtraction angiography, microcatheters, and embolic agents such as polyvinyl-alcohol particles makes superselective embolization of feeding vessels even easier.34
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


