Chapter 26 Basic Mechanisms of Pathological Retinal and Choroidal Angiogenesis
Introduction
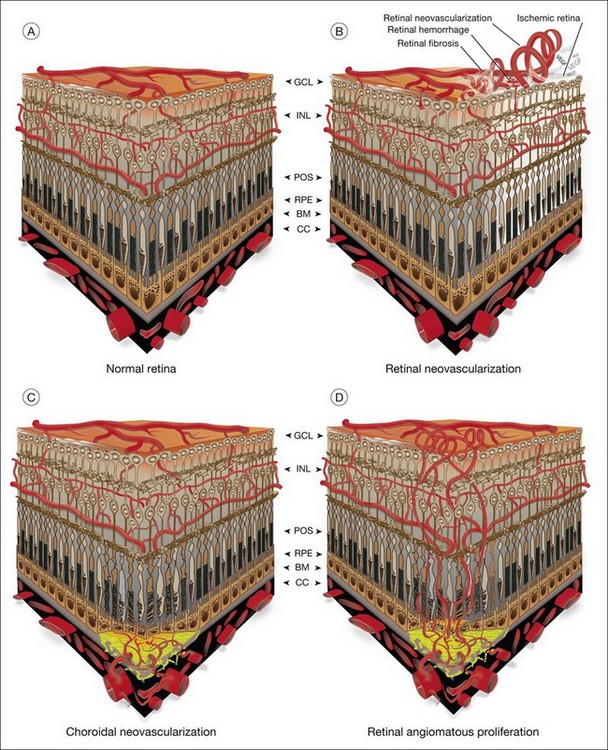
The vasculature of the eye is completely formed shortly after birth and is normally quiescent and nonproliferating in adults.1 An active balance of pro- and antiangiogenic influences is required to maintain this state of homeostasis. Neovascularization, or angiogenesis, occurs when environmental changes result in tissue expression of molecules that favor the growth of abnormal vessels prone to leak, bleed, and cause fibrous proliferation. In the retina, neovascularization causes vitreous hemorrhage and traction retinal detachment in diseases such as diabetic retinopathy (DR) and retinopathy of prematurity (ROP). Choroidal neovascularization (CNV) denotes the pathologic growth of new blood vessels from pre-existing choroidal vessels into the subretinal space. The newly formed vessels lie between the choroid and the retinal pigment epithelium (RPE) or between the native RPE and the neurosensory retina; thus, CNV is also referred to as subretinal neovascularization. In some patients neovascularization forms in the neurosensory retina, and at later stages may extend to the choroidal space. This is also known as retinal angiomatous proliferation or retinochoroidal anastomosis. Neovascularization leads to destruction of the retina and formation of a disciform scar in diseases such as neovascular age-related macular degeneration (AMD) (Fig. 26.1).
Pathogenesis
In angiogenesis, capillary endothelial cells proliferate from pre-existing blood vessels. Activated endothelial cells secrete proteolytic enzymes that dissolve basement membrane around the parent vessel and align themselves to form a new capillary sprout. By curving and elongating, the sprouts form tubes with lumens which anastomose to form loops. Mesenchymal cells are recruited to form smooth-muscle cells, or pericytes, and new basement membrane is deposited.2 More recently it has been demonstrated that hematopoietic stem cells may potentially differentiate into various cellular lineages, including endothelial progenitor cells, which migrate and contribute to sites of choroidal and retinal neovascularization.3–6
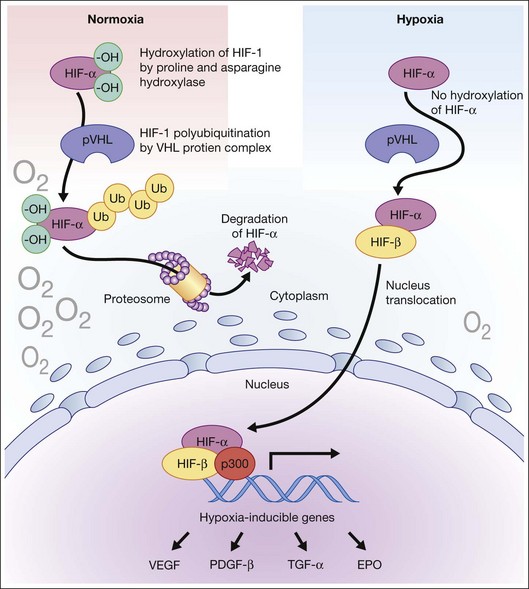
Hypoxia is a primary stimulus for angiogenesis. In early studies of retinal development, growing vessels were noted to invade the developing retina, giving rise to the hypothesis that a diffusible hypoxia-induced growth factor initiated this blood vessel growth.7,8 In DR and other adult vasculopathies, neovascularization was associated with retinal capillary nonperfusion.9–11 Experimental laser vein occlusion in the miniature pig and monkey models have demonstrated this.12,13 Hypoxia prevents the ubiquitin proteosome system degradation of the transcription factor hypoxia-inducible factor 1-alpha (HIF-1α). Stable HIF-1α binds to the promoter region of its major target gene, vascular endothelial growth factor (VEGF), resulting in transcriptional upregulation of VEGF and causing angiogenesis (Fig. 26.2).14–16
Response to tissue injury also plays a key role in angiogenesis. CNV occurs both experimentally and clinically after injury to the outer retina, iatrogenic laser injury, traumatic choroidal ruptures, and outer retinal choroidopathies.17,18 Sublethal cellular injury may release growth factors such as basic fibroblast growth factor (bFGF or FGF-2).19 Growth factor-secreting monocytes may be recruited to the site of tissue injury, which may increase the expression of growth factors causing angiogenesis.20
Free radicals in the form of reactive oxygen species (ROS) or intermediates have been implicated in the pathogenesis of numerous vascular diseases. ROS initiate the repair process by stimulating an angiogenic response. There is evidence that, over time, continual ROS stress depletes tissue of its antioxidant capabilities. Oxidative stress and inflammation may play a role in the pathogenesis of AMD.21 Increase in free radicals is believed to result in an overexpression of growth factors by the RPE, causing a proinflammatory state, damage to Bruch’s membrane, and ultimately CNV.22 The Age-Related Eye Disease Study (AREDS), a multicentered randomized clinical trial, showed a clinical benefit of high-dose antioxidants in reducing visual loss associated with macular degeneration.23
CNV associated with AMD (Fig. 26.3)
Aging and senescence of the RPE
The incidence and progression of nearly every feature of AMD, including CNV, relate to age.24 Lipofuscin, a byproduct of photoreceptor outer-segment digestion by lysosomes, increases with age in RPE as lysosomal activity decreases in RPE with aging. Progressive accumulation of lipofuscin is thought to result in disturbance of RPE function. Aged RPE cells may decrease production of antiangiogenic molecules such as pigment epithelium-derived growth factor (PEDF) as successive passage of cultured RPE cells results in diminished production of PEDF.25–27
Drusen, basal laminar/linear deposit formation
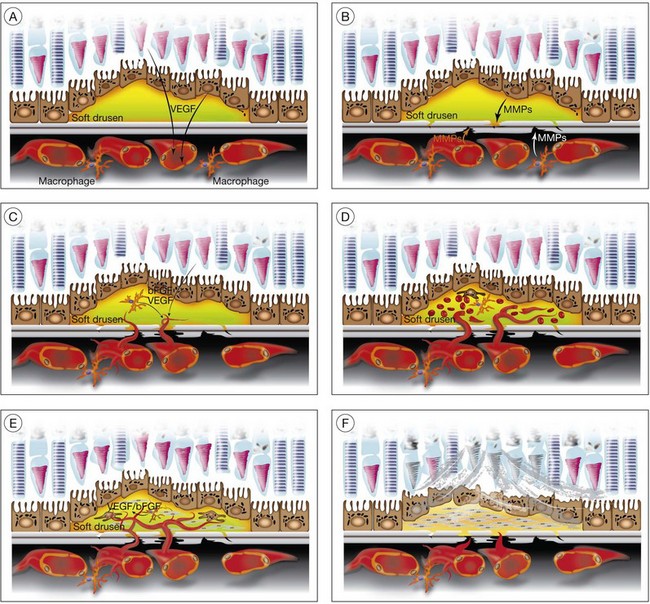
Soft drusen, unlike hard drusen, appear to be an important associated and predisposing feature of CNV. It is thought that membranous accumulation of debris as part of a diffuse disturbance of the RPE, softening of hard drusen, and cleavage in basal laminar/linear deposits may aid in the formation of soft drusen.28–32
Histopathologic studies also reveal that basal laminar deposits (accumulating between the plasma and basement membrane of the RPE) and basal linear deposits (with a thickening of the inner collagenous zone of the Bruch’s membrane) also have important associations with CNV.20 Therefore, abnormal deposits that occur with diffuse distribution pattern between the RPE layer and Bruch’s membrane are predisposing features of CNV. It is hypothesized that deposits between the RPE layer and Bruch’s membrane may block the diffusion of oxygen and nutrients from choriocapillaris to the RPE monolayer and photoreceptors.33 This speculative localized cellular hypoxia could result in overexpression of angiogenic growth factors such as VEGF, which, in turn, induce neovascularization from the choroidal vasculature. Additionally, the deposits are known to contain components of the immune response, and thus may act as initiators of inflammation.34–37 The deposits may also serve as a reservoir for sequestration of factors such as advanced glycation end products (AGE)38,39 that may affect the function of adjacent RPE and choroidal endothelial cells.
Enzymatic and mechanical disruption of Bruch’s membrane
In CNV, activated endothelial cells migrate through Bruch’s membrane; this process occurs by degradation of an intact Bruch’s membrane, or growth through an existing Bruch’s membrane break. Clinicopathologic studies suggest that classic CNVs are predominantly subretinal in location, whereas occult CNVs are predominantly sub-RPE.40 Bruch’s membrane may be disrupted when the balance between proteolytic enzymes such as matrix metalloproteinases (MMPs) and their inhibitors, the tissue inhibitors of metalloproteinases (TIMPs), favors a proteolytic environment. RPE cells express MMP-1 (interstitial collagenase),41 MMP-2 (72-kDa gelatinase),41,42 MMP-3 (stromolysin), and MMP-9 (92-kDa gelatinase),41 as well as TIMP-1,42,43 TIMP-2, and TIMP-3.43 Thus, proteolysis of Bruch’s membrane may potentially result from reduced expression of TIMPs or increased expression of MMPs. MMPs also play an important role in degrading the extracellular matrix at the leading edge of neovascular fronds. It has been shown that decreases in thickness and integrity of the elastic layer of Bruch’s membrane are seen in the macula of eyes with AMD but not in controls.44 Lysyl oxidase-like (LOXL) protein 1 has been shown to guide the spatially defined deposition of elastin and is essential for the maintenance of elastic fibers. LOXL1-deficient mice have been shown to develop increased neovascularization after laser injury.45 Impairment in other components of the Bruch’s membrane such as collagen XVIII has been shown to affect CNV formation in animals. Collagen XVIII knockout mice develop normal choroidal vasculature; however, they demonstrate increased size and leakage of laser-induced CNV.46
Transgenic mice with an intact Bruch’s membrane that overexpress VEGF in photoreceptors develop subretinal neovascularization; however, the subretinal vessels extend from retinal vessels rather than the choroidal vasculature.47 In contrast, transgenic mice that overexpress VEGF in RPE cells show intrachoroidal CNV.48 These findings support the notion that CNV requires both the expression of an angiogenic factor and a break in Bruch’s membrane (by proteolysis, physical disruption, or pre-existing break).
Macrophages are an alternative source of enzymes (such as MMPs) that could cause focal disruption of Bruch’s membrane.49 Histopathologic studies reveal that macrophages accumulate near thinned segments of Bruch’s membrane.50 In AMD, the RPE shows increased expression of monocyte chemoattractant protein-1 (MCP-1), a factor critical for macrophage recruitment.51 Macrophages in the choroid may subsequently degrade Bruch’s membrane, thus forming a passage that can be used by activated choroidal endothelial cells to gain entrance to the sub-RPE space.
Complement, AMD, and CNV
Recent studies suggest that complement dysregulation may mediate AMD pathogenesis. Complement components have been detected in drusen29,30,34–37 and an animal model of CD59 knockout leads to increased CNV.52 Most recently, a Y402H polymorphism in complement factor H (CFH) has been associated with increased risk of AMD and likely wet AMD.53–56 This particular allele of CFH leads to decrease in its activity and thus an increased complement-mediated inflammation since CFH is a negative regulator of the alternative pathway. Other human studies have identified protective haplotypes in two other genes encoding complement proteins, factor B (BF) and complement component 2 (C2).57
Studies of human donor eyes have shown increased levels of C-reactive protein and decreased CFH in the RPE/Bruch’s membrane/choriocapillaris region of eyes with AMD compared to controls, suggesting a role of complement and inflammation in the progression of the disease.58
Inflammation, bone marrow-derived cells, and CNV
Macrophages are an additional source of angiogenic growth factors that may promote the development of CNV. Activated macrophages show increased expression of inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), which may promote angiogenesis by stimulating VEGF expression in RPE.59 Depletion of macrophages diminishes the lesion size and severity in experimental laser-induced CNV.60,61 Ccr-2 knockout mice, characterized by hampered macrophage recruitment, also show inhibition of laser-induced CNV.62
Microglial cells (MC) are involved in phagocytosis of injured and dead cells. Dysfunction of phagocytosis of accumulating debris may lead to further inflammation.63 CX3CR1 is a cytokine produced by MC, and an association between AMD and CX3CR1 has been reported.64–66 A double knockout of MCP1 and CX3CR1 in an animal model leads to an AMD phenotype with spontaneous CNV formation.67,68
The contribution of bone marrow-derived endothelial cells to CNV has been evaluated by inducing CNV in irradiated mice that have received bone marrow transplants from green fluorescent protein (GFP)-expressing mice. These studies show that GFP+ endothelial cells are incorporated into the laser-induced CNV, suggesting that bone marrow-derived progenitor cells provide an additional source of endothelial cells in CNV.6,69,70
CNV membrane formation
In a series of 760 eyes with AMD from 450 patients; 310 eyes (~40%) demonstrated a disciform membrane.20 The mean diameter of these membranes was 3.73 mm, and the mean thickness was 0.27 mm. Preservation of photoreceptor cells was seen when the thickness of the disciform scars was 0.2 mm or less. Histologic studies of highly vascular CNV show that they contain endothelial cell-lined channels, RPE cells, and macrophages within the extracellular matrix-rich stroma. The stromal RPE are immunoreactive for smooth-muscle actin, indicating a transdifferentiated phenotype. The transdifferentiated RPE often expresses VEGF, suggesting that these RPE have a proangiogenic role, and that VEGF is a mediator of AMD-related CNV.71
CNV likely occurs when there is an imbalance between proangiogenic and antiangiogenic growth factors. Angiogenic growth factors, such as VEGF, angiopoietins (Ang 1, Ang 2), and bFGF, released from RPE cells and/or other retinal cells, promote CNV. Choroidal angiogenesis is inhibited by antiangiogenic growth factors such as thrombospondin-1 and PEDF72 and by Fas-mediated killing of new vessels by Fas-ligand-positive RPE or leukocytes.73
Cicatricial membrane formation
Cellular and highly vascularized membranes gradually evolve into paucicellular cicatricial membranes. The loss of cellularity is most likely due to cell death of stromal cells.74 Surgically excised AMD-related CNV contains apoptotic stromal RPE, endothelial cells, and macrophages. Cell death may be associated with a local decrease in the expression of angiogenic growth factors that promote survival of activated cells. Fas and Fas ligand expression may also be involved in the induction of apoptosis in these cells.75 Little is known about the mediators of collagenous scar formation in CNV. Studies have shown that connective tissue growth factor (CTGF), a proangiogenic and profibrotic growth factor, is expressed in stromal RPE cells in surgically excised CNV.76 The development of a cicatricial disciform lesion promotes overlying photoreceptor cell loss.77
Neovascularization associated with diabetic retinopathy
Neovascularization in DR can be thought of as a two-step process. An initial vascular dropout/occlusion phase is followed by abnormal vascular growth. Chronic hyperglycemia leads to vascular injury with basement membrane thickening, pericyte loss, microaneurysms, and vascular leakage.78,79 Many biochemical pathways (protein kinase C, nuclear factor (NF)-κB, mitogen activated protein kinase) have been involved in the pathogenesis of early DR but the exact mechanism remains ill defined. Oxidative injury, microthrombi formation, inflammatory mediator upregulation, and leukostasis have been observed. Proinflammatory cytokines IL-1β and TNF-α are elevated in animal models of DR, recruiting leukocytes and accelerating vascular dropout by promoting further inflammation as well as activating apoptosis directly or indirectly.80–82 This leads to vascular leakage as well as microvascular occlusion. As a consequence of microvascular occlusion there is inadequate supply of oxygen and nutrients for proper cellular metabolism. This leads neurons and supporting astrocytes to secrete proangiogenic factors such as VEGF and insulin-like growth factor (IGF)-1. Clinical studies have substantiated the role of VEGF in DR.83,84 VEGF levels are elevated in patients with proliferative DR (PDR), and although recent reports have not been able to show decreases of VEGF levels in the short term after panretinal photocoagulation,85,86 successful laser retinal ablation decreases them over the long term.84 In addition, several studies have shown the beneficial effects of anti-VEGF intervention in DR.87–90 Before the discovery of VEGF and its important role in DR, hypophysectomy studies had revealed a role of growth hormone (GH) and its associated factors in DR. IGF-1 levels have been shown to be elevated in the vitreous of patients with PDR relative to control eyes.91–93 Although there may be a role for IGF-1 in retinal neovascularization, our understanding of IGF-1 in DR remains unclear.94 IGF-1 may act indirectly via VEGF. Studies of cultured RPE cells demonstrated that adding IGF-1 in vitro increased VEGF mRNA and secreted protein.95 The accumulation of AGE after long-term exposure to hyperglycemia caused an increase in IGF-1 synthesis in human monocytes, suggesting a role for inflammatory pathways.96
Angiopoietins are molecules that bind endothelial Tie receptors and are involved in angiogenesis during development. Vitreous level of Ang 2 and VEGF were significantly higher in patients with PDR than in controls or those with inactive PDR and the levels of Ang 2 correlated significantly with that of VEGF, suggesting an association of Ang 2 and VEGF with angiogenic activity in PDR.97 A nuclease-resistant RNA aptamer that binds and inhibits Ang 2 but not the related Tie2 agonist, Ang 1, inhibited bFGF-mediated neovascularization in the rat corneal micropocket angiogenesis assay, demonstrating that a specific inhibitor of Ang 2 can act as an antiangiogenic agent.98
More recently, a role of erythropoietin (Epo) in DR has been discovered.99 Epo is involved in the generation of red blood cells but has been shown to be synthesized in astrocytes and its receptors have been detected in photoreceptors. Epo has been shown to have neuroprotective effects in various models.100,101 It has also been shown to be proangiogenic. Epo levels are higher in the vitreous of patients with PDR, and inhibition of both Epo and VEGF leads to greater inhibition of neovascularization.99 However, since Epo has neuroprotective effects, its inhibition for neovascularization may come with significant collateral damage.
In addition to elevation of proangiogenic factors, there is also an imbalance of antiangiogenic factors. PEDF, now known as serpin peptidase inhibitor clade F member 1(SERPINF1), is considered to be the most potent inhibitor of neovascularization, inhibiting endothelial cell migration with a median effective dose of 0.4 nM.102 It specifically interferes with neovasculature and not with established vessels due to its mechanism of action through Fas/FasL and its cooperation with angiogenic factors. SERPINF1 upregulates FasL on endothelial cells, whereas angiogenic factors induce its essential partner Fas receptor on neovessels but not on established vessels.103 In PDR, it has been found that PEDF levels are downregulated compared to controls.104 PEDF also has neuroprotective effects, and thus seems to be an ideal candidate for therapeutic intervention.
Inflammation is another important player in the pathogenesis of proliferative DR. Inflammatory cytokines such as TNF-α, IL-1β, intercellular adhesion molecule (ICAM), inducible nitric oxide synthase (iNOS), and IL-6 are elevated in patients with PDR.105–108 Localized inflammation is thought to lead to leukostasis through the interaction of ICAM and CD18.81,109–111 This leukostasis leads to local vaso-occlusion, nonperfusion, and ischemia, which leads to upregulation of angiogenic factors. IL-1β can promote endothelial cell proliferation, propagate inflammation, and upregulate HIF-1α.112,113 TNF-α can promote angiogenesis, and macrophage-induced angiogenesis is mediated through TNF-α.114,115 TNF-receptor p55-deficient animals are protected from retinal neovascularization in animal models.116 Inhibitors of inflammation such as cyclo-oxygenase 2 inhibitors and aspirin have been shown to curtail vascular pathology and neovascularization.81,117,118
The proinflammatory environment is characterized by elevated MMPs, which degrade the extracellular matrix, a step necessary for angiogenesis. Additionally, MMP-9 not only is induced by IL-8119 but also activates IL-8,120 which recruits more inflammatory cells, feeding a destructive positive-feedback loop. Insulin121 and PEDF122 are degraded by MMP-9, leading to a state of more insulin resistance and neuronal peril. Beránek et al. in 2008 showed MMP2 and MMP9 to be elevated in patients with PDR.123
Neovascularization associated with retinopathy of prematurity
Retinal vasculature development begins at about the fourth month of gestation and is complete at about 40 weeks. When an infant is born prematurely, it moves from a hypoxic (Pao2 ~30 mmHg) to relative hyperoxic (Pao2 ~ 60–100 mmHg) environment.124,125 The increase in oxygen tension, coupled with weak antioxidant defenses, leads to production of toxic ROS and decreased HIF. The combination of these effects leads to damage of the vascular endothelial cells and decreased production of VEGF and Epo, with resultant cessation of normal vascular development. As the peripheral avascular retina continues to develop in the absence of a developing vascular bed, it becomes relatively hypoxic and secretes VEGF and Epo at levels above normal physiologic values. Accordingly, the new vascular growth becomes exaggerated, misguided, and abnormal, leading to physical traction and potential retinal detachment. It is important to know that there is no direct evidence of the above-described processes from eyes of premature babies, but it is rather an extrapolation from animal data and other vascular eye diseases. However, the importance of VEGF upregulation in the proliferative phase of this disease has been shown in humans, since blockade of VEGF with anti-VEGF antibodies (bevacizumab) showed significant efficacy in infants with stage 3+ zone I disease.126
Oxygen level changes cannot fully account for disease progression and prematurity is the strongest risk factor for ROP development. Growth factors such as IGF-1 have been implicated by epidemiological and animal studies. Babies with decreased levels of IGF-1 have slower vascular development in the first phase of the disease and animals that lack IGF-1 have substantial reduction in neovascularization in the proliferative phase of the disease.127
Neovascularization in vascular occlusions
Ischemia and subsequent elevation of angiogenic factors are thought to be major and important contributors in neovascularization after central retinal vein occlusion (CRVO). The role of VEGF in particular has been well established. Aiello et al. in 1994 showed increased VEGF levels in the vitreous of patients with active neovascular CRVO;84 in the same year, Miller et al. showed evidence that VEGF is temporally and spatially correlated with ocular angiogenesis in a primate model of CRVO and that blockade of VEGF could inhibit neovascularization.128 Obviously, VEGF is not the only cytokine elevated in CRVO, and elevated levels of VEGF can be seen without neovascularization. Several interleukins have been shown to be elevated in CRVO as well as MCP-1 and ICAM-1.129–131 Additionally, antiangiogenic factors such as PEDF have been shown to be downregulated in CRVO.132 The alterations of cytokines seen in CRVO not only lead to potential neovascularization but are major contributors to vascular leakage and macular edema. Thus far, only anti-VEGF blockade has been studied as a targeted therapy in human patients with CRVO, and several studies have shown significant (but not absolute) success of such a strategy, suggesting that combination therapies may be needed for improved outcomes.133
Neovascularization in uveitis
Retinal neovascularization and CNV in uveitis are well recognized, although uncommon. Ischemia from nonperfusion and inflammation may contribute to the formation of new vessels. Peripheral ischemia and the extent of neovascularization seem to correlate in predominantly ischemic vasculitides such as Eales disease.134 However, neovascularization of the disc has been seen in other uveitic cases without apparent ischemia.135 Given the fact that neovascularization can regress with corticosteroid and other immune-suppressive treatments, it seems that inflammation alone may be sufficient for retinal neovascularization and CNV in this heterogeneous group of diseases.136–138 Nevertheless, VEGF is still a key molecule in uveitis-associated neovascularization, and several studies have shown benefit (albeit partial) of anti-VEGF therapy.139,140
Genetic aspects of neovascularization
Age-related macular degeneration
AMD has a significant genetic component, as there is higher concordance among monozygotic twins compared to dizygotic twins.141 Family members of individuals with AMD are more likely to develop the disease.
The protein ApoE is involved in the regulation of blood lipids, and has been found to be involved in AMD. The ε2 allele of the APOE gene may increase the risk, whereas the ε4 allele may be protective.142 However, other studies find opposite or no association between ApoE and AMD.143–145
The chemokine receptor CX3CR1 and the Toll-like receptor 4 (TLR4) have been implicated in several, but not all, studies as risk factors for progression of the disease.146–149 Conflicting data exist as well on TLR3 as a risk factor for AMD progression.150–153
Convincing genetic association data exist for a locus of chromosome 10q26, which harbors the pleckstrin homology domain-containing protein 1 (PLEKHA1), the ARMS2 gene product of unknown function, and the trypsin-like protease HTRA1. This locus is associated with a seven- to tenfold increased risk of the disease.154–156 In addition, very strong evidence exists for the CFH gene, whereas the single nucleotide polymorphism Y402H can increase the risk of the disease three- to sevenfold.157–159 However, the variation in these genomic regions alone is unable to predict disease development with high accuracy.
Several other studies have implicated various other components of the complement pathway in disease progression. Variations in complement 2, complement factor B, and CFH-related protein 1 and 3 are thought to have protective odds ratios whereas complement 3 may be associated with increased susceptibility.57,160–163
Mutations in other genes have been associated with macular degenerations other than AMD, such as the ABCR gene, a rod-specific ATP-binding cassette (ABC) transporter seen in Stargardt disease.164,165 Mutations in TIMP-3, an inhibitor of proteolysis found within Bruch’s membrane, are seen in Sorsby fundus dystrophy, an autosomal dominant disorder with histologic changes similar to those of neovascular AMD.166–169
Mutation in the vitelliform macular dystrophy (VMD)2 gene that encodes the protein bestrophin has been found in Best macular dystrophy, and mutation in the epidermal growth factor-containing, fibrillin-like extracellular matrix protein (EFEMP1) gene has been seen in patients with malattia levantinese and Doyne honeycomb retinal dystrophy, which are disorders associated with drusen formation.170,171
Diabetic retinopathy
Although the Diabetes Control and Complications Trial and the UK Prospective Diabetes Study demonstrate the beneficial effects of tight glucose and blood pressure control, patients with good glucose control still develop DR. The recent ADVANCE study reported that intensive glucose control to reduce A1c to 6.5% or less had no effect on retinopathy rates.172,173 In addition, it is apparent that some patients with poor control of their disease may not develop DR even over long periods of time, while others develop DR in a short time despite good disease control. This is exemplified in the Joslin Medalist study, which found that a significant number of elderly patients with type 1 diabetes had no evidence of retinopathy despite surviving over 50 years with diabetes.174,175 These results suggest that genetic factors may play a role in the progression of this disease.
Candidate gene approaches have shown fairly consistent associations with genes encoding aldose reductase (ALR2), VEGF, and receptor for AGE (RAGE) in the progression of DR (reviewed by Liew et al.176). Aldose reductase is involved in the metabolism of glucose into sorbitol inside the cells. Sorbitol accumulation leads to osmotic stress and cell injury. Unfortunately, clinical trials with aldose reductase inhibitors failed to show benefit. It is important to note that in these trials no stratification on ALR2 gene polymorphism status was undertaken.177,178 Polymorphism in the VEGF promoter region has been shown to be associated with DR progression and there may be ethnic variation in the “at-risk” haplotype (reviewed by Liew et al.176).
Retinopathy of prematurity
ROP is a multifactorial disease that has many phenotypic similarities with a genetic disease called familial exudative vitreoretinopathy (FEVR). At least four genes have been implicated in FEVR (norrin–FZD4–LRP5–TSPAN12 signaling pathway), and a candidate gene approach has shown that three of them (NDP, FZD4, and LRP5) are mutated in a small percentage of severe ROP patients.179 Association between VEGF, IGF-1 receptor, and angiotensin-converting enzyme gene polymorphisms have been implicated in some (but not all) studies.180–191 Other suggestive associations have been reported between ROP and several candidate genes such as angiotensin II type I receptor, Indian hedgehog, T-box 5, glycoprotein Ibα polypeptide, and cholesterol ester transfer protein, but confirmation is still needed.192
Environmental factors
Age-related macular degeneration
Several modifiable risk factors have been associated with progression of AMD. Amongst them, smoking has been the most consistently reported risk factor, elevating the risk of disease progression by about twofold.193–195 Additionally, a health-promoting diet (rich in seafood, vegetables, and fruits), physical activity, and a nonsmoker status are thought to lead to a 70% reduction in the risk of developing AMD.196 The AREDS study showed that supplementation with vitamins A and C, zinc, and copper in patients with moderate-severity AMD reduces the risk of progression by about 25%.197 Unfortunately, this was not one of the a priori questions set to be answered by the study, so the benefits of this supplementation are not universally accepted. More recently, the Women’s Antioxidant and Folic Acid Cardiovascular Study discovered that supplementation with folate, vitamins B6 and B12 reduced the risk of advanced AMD by 30–40% in women with high-risk cardiovascular factors.198
Diabetic retinopathy
Smoking, hypercholesterolemia, and hypertension are known factors that worsen disease progression. Smoking increases oxidative stress, and may increase TNF levels as well as alter high-density lipoprotein and low-density lipoprotein levels.199,200 Smoking has also been shown to elevate VEGF levels acutely in about one-quarter of diabetic patients.201 Several studies have shown the link between high blood pressure and cholesterol in the progression of DR. Recent studies show that there is greater benefit in reducing the progression of the disease by lowering cholesterol levels compared to lowering high blood pressure.202,203
Retinopathy of prematurity
Besides early birth and reduced birth weight, other factors have been shown to contribute to development of ROP. Excessively high levels of oxygen in incubators, used to save the lives of premature infants, led to an ROP epidemic and the realization that reducing the level of oxygen given to premature babies reduces the incidence of ROP.204,205 Although oxygen levels and ROP are linked, the exact mechanism is not clear. It is possible that the rate of change of oxygenation rather than the absolute level may be more important for disease progression, and there have been reports that return to high oxygen levels for prolonged time with gradual decline to normal oxygen levels may halt and reverse the disease.206–210
Light exposure has also been investigated, but the LIGHT-ROP study concluded that a reduction in ambient light exposure does not alter the incidence of ROP.211
The diet of premature babies is usually poorer in omega-3 fatty acids compared to mother’s milk. Omega-3 fatty acid supplementation in animal models of ROP has shown reduction in neovascularization.212 Human studies with docosahexaenoic acid supplementation of infant formula at 0.32% of total fatty acids showed improved visual acuity as measured by visual-evoked potential, but this study did not investigate neovascularization.213
Angiogenic and antiangiogenic factors in neovacularization
Vascular endothelial growth factor
VEGF was first identified in tumor models as a vasopermeability factor and initiator of angiogenesis upregulated by hypoxia. In addition to being the strongest endothelial cell mitogen, VEGF has been shown to induce the expression of plasminogen activators in microvascular endothelial cells, which is important in the extracellular proteolysis necessary for capillary formation.214 VEGF induces the expression of endothelial cell α1β1 and α2β1 integrins, which are important in migration.215 There is also in vitro evidence that VEGF upregulates endothelial cell fenestrations in kidney glomerulus, choroid plexus, and even the choriocapillaris.216 Leukocyte adhesion has been shown to be important in vascular leakage. VEGF increases the expression of ICAM-1 on endothelial cells, resulting in increased leukostasis, which mediates the breakdown of the blood–retinal barrier.217,218
VEGF, also known as VEGF-A or VEGF-1, is expressed as five mRNA splice variants in humans: isoforms 121, 145, 165, 189, and 206.219 VEGF is a heparin-binding dimeric glycoprotein with disulfide-linked subunits, which share significant sequence homology with the A and B chains of platelet-derived growth factor (PDGF).220 VEGF 121 is entirely soluble and unbound to extracellular matrix. VEGF 165 is intermediate and binds somewhat to extracellular matrix. VEGF 189 is almost entirely sequestered to extracellular matrix and cell surface sites.221,222 VEGF 165 is the predominantly expressed isoform when human cDNA libraries are screened, and is optimal for bioavailability and biologic potency. It is the critical isoform for both developmental and pathologic retinal angiogenesis.222–224 Two other related endothelial growth factors, VEGF-B and VEGF-C, have structural homology to VEGF and appear to play roles in tumor angiogenesis and in the development of the lymphatic system, as does VEGF-D.225 VEGF-C, also known as VEGF-2, has been shown to promote angiogenesis in the rabbit ischemic hind limb model.226 VEGF-E has similar angiogenic activity to VEGF-A, primarily through VEGF receptor 2.227
Both high- and low-affinity VEGF receptors have been identified on not only endothelial cells, but also on bone marrow-derived and retinal epithelial cells.228–230 They belong to the family of tyrosine kinases requiring phosphorylation to be activated upon ligand binding. VEGFR-1 (FMS-like tyrosine kinase or FLT-1 in human) and VEGFR-2 (fetal liver kinase 1 or Flk-1 in the mouse; TKR-C in the rat; kinase insert domain receptor or KDR in the human) are expressed on endothelial cells, whereas VEGFR-3 (FLT4) is primarily found on lymphatic endothelial cells.231 Although VEGF binds to both VEGFR-1 and VEGFR-2, the latter is primarily responsible for endothelial cell mitogenesis, survival, and permeability.232 VEGFR-1 may be important in development by sequestering VEGF, preventing it from interacting with VEGFR-2.233 VEGFR-1 has an established role in monocyte chemotaxis.234,235 VEGF-C has also been shown to be a ligand for VEGFR-2 and VEGFR-3.226 Synergism between FGF-2 and VEGF has been demonstrated by the finding that FGF-2 increases VEGFR-2 expression in microvascular endothelial and aortic endothelial cells.236 In addition to the receptor tyrosine kinases, VEGF also interacts with a family of coreceptors, the neuropilins, which may enhance its angiogenic function.237
VEGF is expressed by retinal cells in vitro and is upregulated by hypoxia.238,239 Other modulators of VEGF expression are hypoglycemia,234 beta-estradiol, and ROS.240–242 Several growth factors cause upregulation of VEGF gene expression, including epidermal growth factor, transforming growth factor (TGF)-α and β, IGF-1, FGF, and PDGF, implicating both paracrine and autocrine release of these factors in the hypoxic regulation of VEGF.228,243
In vivo work has demonstrated VEGF to be spatially and temporally correlated with iris neovascularization in a monkey model of ischemic retinopathy and iris neovascularization.128 VEGF protein levels in serial aqueous samples have been shown to correlate with the severity of induced retinal ischemia and iris neovascularization. In situ hybridization identified the inner retina as the source of VEGF.128,244 Work in models of ROP also implicates the importance of VEGF in the development of retinal neovascularization.245–247
In vitro, VEGF has been shown to be sufficient to produce neovascularization. VEGF results in neovascularization in the corneal micropocket and in chick chorioallantoic membrane (CAM) bioassay.248,249 A single intra-arterial bolus of VEGF was sufficient to stimulate angiogenesis and collateralization in a rabbit ischemic hind limb model, leading to studies of the therapeutic use of VEGF in peripheral vascular and coronary artery disease.250 Injections of recombinant human VEGF in normal monkey eyes led to iris neovascularization and neovascular glaucoma, as well as many of the changes of DR, including vessel dilation, tortuosity, microaneurysm formation, hemorrhage, edema, capillary dropout, and intraretinal neovascularization.251,252 However, VEGF alone may not be sufficient in vivo to induce retinal neovascularization.
To demonstrate the causal role of VEGF in neovascularization secondary to ischemia, VEGF activity was specifically blocked using anti-VEGF antibodies, soluble VEGF receptors, or antisense oligonucleotides to VEGF. Intravitreous injection of anti-VEGF antibodies completely prevented the development of iris neovascularization in the monkey model.253 In a mouse ROP model, dominant-negative VEGF receptors and VEGF antisense oligonucleotides substantially decreased retinal neovascularization.254,255 This work has confirmed the central role of VEGF in pathologic retinal neovascularization.
VEGF also plays an important role in the development of CNV. Immunostaining of surgical specimens of choroidal neovascular membranes showed increased VEGF expression.71,256 In situ hybridization studies have demonstrated a correlation between VEGF expression and the development of CNV in laser injury models in the rat and monkey.257 Oxidative stress may stimulate the overexpression of growth factors from the RPE, a possible inflammatory state, and subsequent damage and thickening of Bruch’s membrane from recruited macrophages.21,22 It has been suggested that the abnormally thickened Bruch’s membrane may interfere with polarized RPE secretion of VEGF necessary for maintenance of the choriocapillaris. Atrophy of the choriocapillaris, often seen clinically, may result in a state of outer retinal hypoxia, stimulating VEGF-induced angiogenesis.258 Studies have also shown that despite the absence of hypoxia in the laser injury models, specific compounds that bind VEGF and its receptors virtually eliminate CNV.259,260 These data have important clinical implications, as specific pharmacologic inhibitors of VEGF have been approved for the treatment of neovascular AMD and shown to be effective.261–264
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


