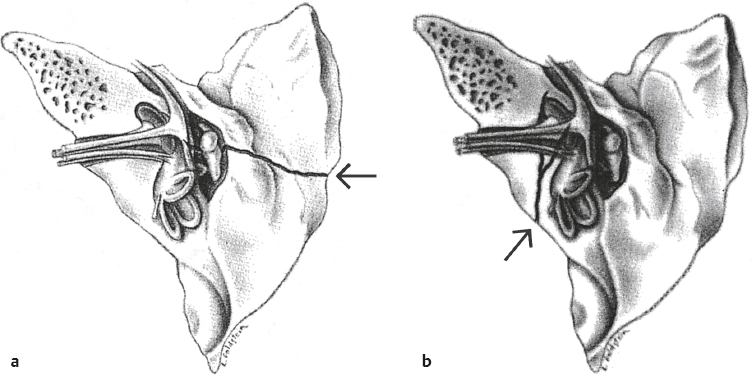
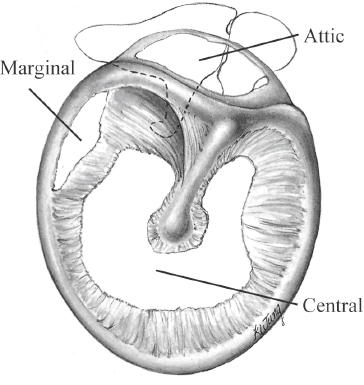
6 Auditory System and Related Disorders This chapter gives an overview of the disorders of the auditory system. We will address the nature of various pathologies, where and when they occur, their major signs and symptoms, how hearing is affected, and the ways they are treated. Further coverage of auditory disorders, their diagnosis, and their treatment may be found in many fine otolaryngology texts (e.g., Hughes 1985; Buckingham 1989; Bluestone, Stool, & Kenna 1996; Paparella, Shumrick, Gluckman, & Meyerhoff 1991; Tos 1993, 1995; Hughes & Pensak 2007; Lalwani & Grundfast 1998; Wetmore, Muntz, & McGill 2012; Van De Water & Staecker 2005).1 Hearing impairments are caused by abnormalities of structure and/or function in the auditory system, which are often called lesions. Using this terminology, a hearing loss may be viewed as one of the manifestations of a lesion somewhere in the ear, as are other symptoms such as pain, ringing in the ears, and dizziness. We are interested in the nature of the lesion, as well as its severity, etiology (cause), location, and time course (including when it began and how it has progressed). Disorders are often called idiopathic if a specific underlying cause cannot be identified. The interactions among these various factors can often be important. Consider, for example, the distinction between “congenital” and “hereditary.” A disorder is congenital if it is present at birth, a matter of timing. On the other hand, a disorder is hereditary or genetic if it is transmitted by the genetic code that the child inherits from her parents; otherwise it is acquired, which is a matter of causation. A congenital disorder may be caused by a genetic problem or other factors that interfere with normal embryological development or occur during the birth process. Similarly, genetic disorders are often present at birth, but others are delayed, manifesting themselves long after birth. Moreover, some degree of hereditary predisposition is involved in many acquired disorders. We are equally interested in the nature, severity, and time course of the hearing impairment itself. Although the nature of a hearing loss goes hand-in-hand with that of the lesion, the same cannot always be said about severity and time course. For example, middle ear infections cause conductive losses, but the magnitude of the hearing loss is not clearly related to the severity of the infection. Similarly, hair cell damage due to noise exposure and/or aging is typically underway long before the patient notices (or at least admits to) a hearing problem. A more dramatic issue has to do with when a severe hearing loss develops in a child and when it is identified and addressed. Prelingual impairments occur before the development of speech and language, and have a catastrophic effect on this process, precipitating serious communicative impairments and interference with academic development. Postlingual losses develop after speech and language have been established, and have a relatively smaller effect. The earlier the onset, and the longer the child is deprived of auditory stimulation, the more the loss will interfere with speech and language development, and hence the more devastating its effect. Conversely, the earlier that a significant hearing impairment is identified, the sooner its effects can be mitigated by appropriate intervention techniques. 1 These texts served as references for material used throughout this chapter. The case history includes information about the patient that provides insight into his auditory status and related factors, and that contributes to the development of a diagnostic impression, a plan for audiological remediation, and appropriate referrals to other professionals. Thus, the clinical case history involves obtaining a complete picture of the patient’s auditory and communicative status, historical information about factors known to influence or to be related to auditory functioning, and his pertinent medical and family history. These points should be kept in mind while reading this chapter, and necessitate addressing the issue of case histories at this point. Exactly how the case history is obtained is often a matter of personal style and interviewing skills. At one extreme is the use of a formal “case history form” that the patient completes in advance, which is then reviewed and discussed with the patient. The other extreme involves conducting an open-ended interview, in effect asking, “What’s a nice person like you doing in a place like this?” This method is quite effective in the hands of a “master clinician,” but it is easy for those with less experience to lose control of an open-ended interview, or to omit information that would have been obtained with a more structured approach. For this reason, those who prefer the open-ended approach are often well served by completing items on a prepared form during the interview rather than starting with a blank sheet of paper. Many audiologists prefer to use a structured interview in which they ask the patient a predetermined set of questions, and then probe further depending on the answers. Regardless of one’s approach to the clinical interview, it is desirable for the evaluation process to include a functional assessment (self-assessment) scale completed by the patient and/or a parent or spouse (see Chapter 16). A “case history form” is not included here because what is covered in a clinical case history should stem from an integrated knowledge and understanding of the nature, signs, and symptoms of auditory and related disorders. The case history can be meaningful only if one has an integrated knowledge and understanding of normal auditory functioning and the characteristics of auditory and related disorders. Otherwise, one is filling out the form for the sake of filling out the form. The serious student will find that an instructive and useful exercise is to derive a case history outline on the basis of the material in this chapter and elsewhere in the text, particularly Chapters 12 and 13. Sensorineural lesions involve the cochlea and/or auditory nerve, and may affect sensory receptor (hair) cells, auditory neurons, and/or any of the many structures and processes that enable them to be activated and function properly. The resulting impairment of auditory functioning is called a sensorineural hearing loss. Nerve deafness and perceptive loss are obsolete terms for sensorineural hearing loss, but are occasionally encountered. Using the term sensorineural (sometimes sensori-neural or neurosensory) highlights the anatomical and physiological interdependence of the cochlea and the eighth nerve. In addition, cochlear and auditory nerve lesions cannot be distinguished on the audiogram because both result in threshold shifts that are the same for air and bone-conduction (i.e., no air-bone-gaps). Moreover, sensory and neural lesions can coexist because, for example, the absence of cochlear hair cells can result in degeneration of the auditory neurons associated with them, and an eighth nerve tumor can indirectly damage the cochlea by putting pressure on its blood supply. In spite of these points, it is not uncommon to find that sensorineural loss is being used to mean or imply “sensorineural loss of cochlear origin,” in which case the intended meaning is usually understood from the context. Disorders of the eighth nerve are often described as being retrocochlear, meaning “beyond the cochlea.” Let us review some of the more common characteristics of sensorineural losses, which are due to cochlear lesions in the preponderance of cases. We will then go over conductive impairments, so that the student can appreciate the major differences between these two broad categories. The characteristics associated with neural lesions, per se, will be covered later in the section on retrocochlear disorders. Cochlear disorders result in a loss of hearing sensitivity that is essentially the same for air and bone-conduction. There is a rather systematic relationship between where a lesion occurs along the cochlear spiral and which frequencies have elevated thresholds on the audiogram: High-frequency hearing losses are associated with damage toward the base of the cochlea, and the hearing loss includes successively lower frequencies as the damage to the cochlea extends upward toward the apex. Similarly, lesions affecting the apical regions of the cochlea are associated with low-frequency sensorineural hearing losses, and the loss widens to include successively higher frequencies as the cochlear abnormality spreads downward toward the base. The outer hair cells are generally more susceptible to damage than the inner hair cells, and lesions involving the outer hair cells alone are typically associated with mild to moderate degrees of hearing loss. Damage to both the outer and inner hair cells produces more severe losses. Locations along the basilar membrane without functioning inner hair cells (and/or auditory nerve fibers) cannot respond to stimulation, and are known as dead regions (Moore 2004). However, it is difficult to establish a clear relationship between the severity of cochlear damage and the amount of hearing loss it causes. Unlike the situation for many conductive disorders, medicine and surgery cannot correct sensorineural hearing losses because they are due to cochlear and/or neural damage that is permanent. In effect, missing hair cells and neurons do not regenerate. The student should be aware that hair cell regeneration can occur in birds (Corwin & Cotanche 1988; Ryals & Rubel 1988; Cotanche, Lee, Stone, & Picard 1994; Tsue, Oesterle, & Rubel 1994). This is an avenue of critically important research that might provide hope for the future. However, counseling skill is often needed to help patients be aware (and accept) that while research on hair cell regeneration and related issues is critically important, it is also unrealistic to expect practical benefits for hearing improvement within the foreseeable future. Sensorineural hearing losses can be of any shape and degree, but the most common configurations have thresholds that get worse as frequency increases. In other words, sensorineural impairments often involve a greater loss of hearing sensitivity at higher frequencies than at lower frequencies, so that the audiometric configuration may be described as sloping. This creates a burdensome problem for the hearing-impaired patient because many of the acoustical cues that distinguish speech sounds involve the higher frequencies, and because, on average, the intensity of the speech signal gets weaker as frequency increases. Most patients with sensorineural hearing losses complain that they can hear speech, but that it is unclear or hard to understand, and that this problem becomes worse when noise or competing sounds are present. These ubiquitous complaints occur for several reasons. Many high-frequency speech cues are rendered inaudible or barely audible by the frequency dependence of many of the sensorineural losses described previously. In addition, speech cues are distorted because inner ear lesions cause a variety of auditory impairments above and beyond elevated thresholds, such as the dulling of fine frequency and temporal distinctions. As a result, the auditory representation of speech cues is less faithfully encoded and noisy. In other words, cochlear lesions impair the clarity of speech due to both attenuation and distortion (e.g., Plomp 1986; Vermiglio, Soli, Freed, & Fisher 2012). Patients with cochlear disorders often experience aberrations of pitch and loudness perception, such as diplacusis and loudness recruitment. Diplacusis means that more than one pitch (a perception) is heard in response to the same pure tone (i.e., frequency). This phenomenon is called monaural diplacusis when the same tone elicits different pitch sensations within the same ear, and binaural diplacusis when there is a pitch difference for the same tone between the two ears. Loudness recruitment means that the loudness of a sound (a perception) grows abnormally rapidly as the intensity of the sound (its physical level) is raised above the patient’s threshold. For example, suppose a patient has a 50 dB HL cochlear loss in one ear and normal hearing (0 dB HL) in the other ear. A 100 dB HL sound would be 100 dB above the threshold in the normal ear but only 50 dB above threshold in the abnormal ear; yet 100 dB would sound equally loud in both ears. In other words, a 50 dB increment in the impaired ear sounds as loud as the 100 dB increment in the normal ear. As a result, many sounds are either too soft to hear adequately or too loud to hear comfortably. This is why a hearing-impaired patient might ask you to “speak up” and then ask you to “stop shouting” after you comply with the first request. It also creates a dilemma for hearing aid use because the same amount of amplification that makes softer sounds audible also makes more intense sounds too loud. Patients with severe and profound degrees of sensorineural hearing loss will not be able to hear speech without amplification. The inability of these patients to monitor their own speech can lead to aberrations in vocal pitch and loudness, as well as articulation errors. Along with language disorders, speech production problems are a major issue for patients with prelingual hearing losses, but are less common than once thought among adults with adventitious impairments. Conductive lesions impair the transmission of sound from the environment to the cochlea, so that the signal reaching the sensorineural system is weaker than it should be. A conductive hearing loss is the amount by which the signal is attenuated (weakened) due to the disorder, and is expressed by the size of the air-bone-gap (Chapter 5). For example, a 30 dB conductive hearing loss means that the signal reaching the cochlea is 30 dB weaker than it would have been if the conductive mechanism had been normal, resulting in a 30 dB HL hearing loss. Assuming that the patient has a normal sensorineural system (with a bone-conduction threshold of 0 dB HL), this 30 dB conductive loss would cause (1) a 40 dB HL signal to sound softer than normal because the signal reaches the cochlea at 40 – 30 = 10 dB HL, and (2) a 25 dB HL signal to be inaudible because it reaches the cochlea below threshold at 25 – 30 = –5 dB HL. The size of the conductive hearing loss is not necessarily related to the severity of the underlying disease (e.g., an ear infection), but rather depends on how the lesion impedes the transmission of energy to the cochlea (e.g., by interfering with the middle ear transformer function). In contrast to the sloping configuration of the “typical” sensorineural impairment, conductive losses as a group tend to have relatively little variation in the amount of hearing loss from frequency to frequency. Notice that the emphasis is on the word relatively, and that conductive audiograms do not have to be “flat” any more than sensorineural audiograms have to be sloping; there are many exceptions to both generalities. Because conductive disorders affect energy transmission to the cochlea but not the sensory processes within the cochlea, we expect the patient’s complaints to reveal that she is experiencing a loss of intensity but not distortions or a loss of clarity; for example, “Speech is too soft but it does sound clear once it’s loud enough.” However, patients do not necessarily characterize their perceptual experiences using our concepts. Thus, when the patient says something like “Speech sounds muffled,” we need to find out what she means by “muffled.” Does it mean that speech is too low unless people speak loudly (a typical conductive loss complaint) or that speech is lacking in clarity even though it is loud enough (often associated with sensorineural loss)? Relatively intense sounds are generally less bothersome to patients with conductive hearing losses because these sounds are reduced in level before entering the cochlea. In one sense, the conductive loss is like an “ear plug” that lowers sounds by the amount of the air-bone-gap. For example, a 100 dB HL sound will reach the cochlea of a patient with a 40 dB conductive loss (air-bone-gap) at only 60 dB HL (i.e., 100 – 40 = 60). In contrast, the same 100 dB HL sound would reach a normal person’s cochlea at 100 dB HL, and thus be very loud. This phenomenon is advantageous when these patients use hearing aids because it permits many sounds to be amplified without becoming too loud. However, this apparent “benefit” of a conductive hearing loss is no larger than the size of the air-bone-gap, and certainly does not shield the ear from sounds that are intense enough to cause a tactile sensation or pain. Patients with conductive losses may report that speech is heard better in a noisy environment than in a quiet one. This phenomenon is the opposite of the normal experience and is called paracusis willisii. It occurs for the following reasons: Normal people have no trouble hearing conversational speech in a quiet room, but this speech is too low for the patient with a conductive loss. People talk louder in the presence of a background noise because of the Lombard voice reflex (see Chapter 14). However, the background noise level is still high enough to interfere with the ability to hear the talker’s voice for normal-hearing people. In contrast, the background noise is effectively lowered (or even made inaudible) by the conductive hearing loss, while at the same time the talker’s increased vocal effort makes his speech intense enough to become audible above the noise for people with conductive losses. In other words, the noise makes the talker speak louder, and the conductive loss makes the noise sound lower. It is also common (but certainly not universal) to find that patients with conductive losses speak relatively softly. This occurs because the patient hears her own speech loud and clear via bone-conduction (which is normal) butat the same time fails to adjust her vocal level to account for environmental noises that are rendered inaudible or very low due to the hearing loss. A mixed hearing loss is the combination of a sensorineural loss and a conductive loss in the same ear. Mixed losses may be caused by the presence of two separate disorders in the same ear (e.g., noise-induced hearing loss plus otitis media) or by a single disorder that affects the conductive and sensorineural systems (e.g., head trauma or advanced otosclerosis). Tinnitus is the abnormal perception of sounds for which there is no external stimulus (see, e.g., Tyler, 2000 2006; Snow 2004; Henry, Dennis, & Schechter 2005). Tinnitus is typically associated with a wide variety of sensorineural and conductive hearing losses, but it also occurs when hearing is within normal limits. The sensations are often described as ringing in the ears, head noises, or ear noises, and the sounds are variously characterized as tonal, ringing, buzzing, rushing, roaring, hissing, chirping, pulsing, humming, etc. Sounds audible only to the patient are called subjective tinnitus, in contrast to objective tinnitus (or somatosounds), which also can be detected by an examiner, and is much less common. A particular etiology for subjective tinnitus is evident in some cases (e.g., Meniere’s syndrome and acoustic neuroma), but an identifiable original source is not clear in most patients with chronic tinnitus. Moreover, while most cases of chronic tinnitus are initiated by damage to the cochlea, its persistence and the annoyance it causes are related to processes and changes in both auditory and nonauditory aspects of the central nervous system (Henry, Roberts, Caspary, & Theodoroff 2014). Objective tinnitus tends to be associated with sources involving, for example, the vascular system, Eustachian tube, temporomandibular joint, and/or muscular activity. As a result, medical assessment is an important step in the assessment of the patient with tinnitus. Chronic tinnitus may be the most common health-related complaint, affecting ~ 10 to 15% of the population, and having symptoms severe enough to affect day-to-day living in roughly 1 to 2.5% (e.g., Davis & El Refaie 2000; Henry et al 2005; Shargorodsky, Curhan, & Farwell 2010; Kochkin, Tyler, & Born 2011). To appreciate the impact of clinically significant tinnitus, let us consider some statistics derived from a database by Meikl, Creedon, & Griest SE (2004) on 1630 patients seen for tinnitus management. Sixty-nine percent of their patients reported that their tinnitus caused moderate or greater degrees of discomfort, 74% were uncomfortable in quiet environments, and 92% had difficulty ignoring their tinnitus. Sleep interference was a problem for 71% of their patients, 82% reported irritability or nervousness, 82% had difficulty relaxing, and 79% had difficulty concentrating. Moderate or greater degrees of interference were experienced by 62% for social activities, 52% for work activities, and 72% for overall enjoyment. As many as 45% of patients with tinnitus also experience hyperacusis (Henry et al 2005); conversely, roughly 86% of those with hyperacusis also experience tinnitus (Anari, Axelsson, Eliasson, & Magnusson 1999), and the evaluation and treatment of these problems are intertwined (see Chapter 15). Hyperacusis refers to an intolerance for sound in which the patient experiences discomfort, and is often distinguished from misophonia and phonophobia, although these terms are variously defined (e.g., Vernon 1987, 2002; Katzenell & Segal 2001; Jastreboff & Jastreboff 2002; Baguley 2003; Jastreboff & Hazel 2004). Misophonia and phonophobia implicate emotional components in one’s reactions to sounds, with the former involving a dislike of sounds and the latter connoting actual fear. The information needed to make a particular person (the genetic code) exists in the form of deoxyribonucleic acid (DNA) molecules packaged as chromosomes in every cell. Genes are segments of DNA that occur at fixed locations on the chromosomes, and operate as the biological units of inheritance. In other words, hereditary characteristics (traits) are carried by genes, which are arranged along the chromosomes. Humans have 46 chromosomes arranged in 23 pairs. One pair is different for females (XX) and males (XY), and the other 22 pairs are autosomes, or the same for both sexes. Each parent contributes half of each pair. A genetic or hereditary disorder is an abnormal trait that is transmitted by an abnormal gene. Disorders may be the result of single- or multiplegene abnormalities, or multifactorial inheritance, which is the combined effect of genetic and environmental factors. Hereditary hearing losses can occur alone (nonsyndromic) or in combination with other genetic abnormalities (syndromic), and may be transmitted by autosomal dominant, autosomal recessive, and X-linked inheritance, and by mitochondrial mutations (e.g., Friedman, Schultz, Ben-Yosef, et al 2003; Fischel-Ghodsian 2003; Nance 2003; Van Laer, Cryns, Smith, & Van Camp 2003; Smith, Shearer, Hildebrand, & Van Camp 2013; Toriello & Smith 2013; Van Camp & Smith 2013; OMIM 2013). Genetic hearing losses occurring alone are called nonsyndromic, and we shall see that they have been associated with a large number of specific genes and/or chromosome locations of the genes in the human genome. (Syndromes involving hearing loss are discussed later in this chapter.) Autosomal dominant inheritance means that only one abnormal gene is needed in order for the trait to appear (hence, it is dominant). Suppose one parent is hearing impaired due to an autosomal dominant gene and the other parent is normal. It does not make a difference which parent is affected because the sperm and egg each contribute half of every chromosome pair. If the mother is affected, then there will be a 50% chance that a given egg will contain the abnormal gene. If the father is affected, then half of his sperm will carry the abnormal gene and half will not. In either case, the chances are 50-50 for any pregnancy to result in a hearing-impaired child, as illustrated by the pedigree diagram in Fig. 6.1a. The situation is different with autosomal recessive inheritance because both genes in the pair must be present for the trait to manifest itself. There are three possibilities with respect to the abnormal recessive gene: (1) One may be altogether free of the abnormal gene. (2) A person with both abnormal genes will have the disorder and will be able to transmit the disorder to an offspring. Such an individual is a homozygote because the same (homo) kind of abnormal gene was contributed by both parents when the fertilized egg (zygote) was produced. (3) An individual may have only one abnormal recessive gene, while the other one in the pair is normal. This person is a heterozygote because the two genes in the pair are different (hetero). He will not have a hearing loss because he has only one abnormal gene and it is recessive, but he is a carrier because he can transmit the abnormal gene to his offspring. Let’s see what happens when both parents are normal-hearing carriers of a certain recessive gene for hearing loss, that is, heterozygotes with one abnormal gene and one normal gene (Fig. 6.1b). The probability of getting the mother’s abnormal gene is 0.5 and the probability of getting the father’s abnormal gene is also 0.5. Therefore, the probability of getting both abnormal genes is 0.5 × 0.5 = 0.25. In other words, a given pregnancy has a 25% chance of producing a child who has both abnormal genes and is hearing impaired. On the other hand, the probability of getting both normal genes is also 0.5 × 0.5 = 0.25, which means a 25% chance that a given pregnancy will result in a child who is altogether free of the abnormal gene. What about the other 50%? These are the two remaining combinations (normal gene from the mother with abnormal from the father, and abnormal from the mother with normal from the father), both of which produce an unaffected carrier of the abnormal gene. Autosomal recessive hearing losses can be very hard to trace because a particular kind of abnormal gene can be transmitted across several generations before two unaffected (and unaware) carriers coincidentally find each other and fail to beat the odds. Fig. 6.1 Pedigree diagrams for disorders based on (a) autosomal dominant, (b) autosomal recessive, (c) X-linked recessive, and (d) X-linked dominant inheritance. Circles, females; squares, males; open symbols, individuals free of the abnormal gene; filled symbols, affected individuals with the abnormal gene; “C,” unaffected carriers of the abnormal gene. X-linked (or sex-linked) inheritance occurs when the gene is associated with the X chromo-some instead of being autosomal. With an X-linked recessive disorder (Fig. 6.1c), an affected male transmits the abnormal gene to all of his daughters, who become unaffected carriers of the trait, but not to his sons. In turn, a female carrier has a 50% chance of having sons who are affected and a 50% chance of having daughters who are unaffected carriers. In X-linked dominant inheritance (Fig. 6.1d), an affected male will have all affected daughters and no affected sons. An affected female has a 50% chance of having affected children, whether they are males or females. In contrast to the DNA in the nucleus, which is inherited from both parents, the 37 genes of the mitochondrial DNA are inherited from the mother alone. Mitochondrial mutations cause both syndromic and nonsyndromic deafness, and the latter is often related to aminoglycoside ototoxicity (e.g., Fischel-Ghodsian 2003; Kokotas, Petersen, & Willems 2007; Forli, Passetti, Mancuso, et al 2007; Bindu & Reddy 2008; Pandya 2011; Van Camp & Smith 2013). Ototoxicity is discussed later in this chapter. About 50% of prelingual hearing losses ≥ 40 dB are hereditary, of which roughly 70% are nonsyndromic (Smith et al 2013). Approximately 75 to 85% of the nonsyndromic hearing losses are autosomal recessive, 15 to 20% are autosomal dominant, and 1 to 2% are X-linked. Mitochondrial defects account for less than 1% of the cases. To date, at least 26 genes have been linked to autosomal dominant deafness, 36 genes to autosomal recessive deafness, 4 to X-linked deafness, and 7 to mitochondrial losses (Smith et al 2013; Van Camp & Smith 2013; OMIM 2013).2 These hearing losses are identified by DFN (for “deafness”) followed by A for dominant, B for recessive, or X for X-linked, and then a number. For example, DFN1 is an autosomal dominant hearing loss involving the DIAPH1 gene; DFNB21 is an autosomal recessive hearing loss associated with the TECTA gene, and DFNX1 is an X-linked hearing loss related to the PRPS1 gene. 2 Research in this area is progressing very actively. For up-to-date information and links to the current literature, see http://hereditaryhearingloss.org/, http://www.nhgri.nih.gov/, and http://www.omim.org/. The most common form of genetic hearing loss is DFNB1, which accounts for half of the autosomal recessive cases (Smith et al 2013; Van Camp & Smith 2013). Children born with DFNB1 have prelingual bilateral sensorineural hearing losses that can vary in degree of severity from mild to profound, and are nonprogressive. It is caused by mutations of the GJB2 and GJB6 genes, which provide for the expression of proteins in the inner ear called connexin 26 and 30. The connexins form gap junctions, which are channels that permit substances such as ions to flow across cell membranes, and are thus essential for maintaining the required balance and flow of potassium ions and other functions in the inner ear; however, the exact mechanism of the hearing loss is not fully understood (see, e.g., Ortolano et al 2008; Nickel & Forge 2008, 2010; Toriello & Smith 2013). Approximately 30% of significant hereditary hearing losses occur in syndromes (Smith et al 2013). A syndrome is a pattern of abnormalities and/or symptoms that result from the same cause. Related terms include an association, which describes a group of abnormalities that occur together too often to be due to chance; and a sequence, which is a group or pattern of abnormalities that result from a primary anomaly. Auditory disorders can be found in a great many syndromes, and extensive listings are readily available (e.g., Northern & Downs 1991; Hall, Prentice, Smiley, & Werkhaven 1995; Friedman et al 2003; Toriello & Smith 2013; Van Camp & Smith 2013; OMIM, 2013). Several representative syndromes known to cause hearing impairments are outlined in Table 6.1. Notice that different syndromes are associated with different kinds of hearing loss; some appear to be present at birth (congenital), whereas others are delayed in onset. Other syndromes are discussed elsewhere in the chapter. Fetuses and newborns are adversely affected in various ways by at least 16 viruses and 6 bacteria. The major offenders are called the TORCH complex, which includes toxoplasmosis, “other” (including syphilis), rubella, cytomegalovirus, and herpes simplex. Many authorities have replaced TORCH with STORCH or (S)TORCH to recognize the importance of congenital syphilis in this group. Syphilis (lues) is a sexually transmitted bacterial infection caused by the spirochete Treponema pallidum, which can be passed to the fetus from an infected mother. The number of reported cases of congenital syphilis increased dramatically from only 160 in 1981 to 2867 in 1990 (Shimizu 1992). Congenital syphilis is associated with notched incisor teeth, interstitial keratitis (a chronic inflammation of the cornea with the appearance of ground glass), and sensorineural hearing loss. The hearing loss can develop at any time during childhood or adulthood, even as late as ~ 60 years old. The sensorineural hearing loss is usually bilateral, symmetrical, and progressive, typically becoming severe-to-profound in degree. It is not uncommon for the loss to have a sudden onset or to fluctuate over time. Vertigo and/or tinnitus can also occur. Vertigo is a specific kind of dizziness in which the patient experiences a sensation of whirling or rotation. It is associated with nystagmus (Chapter 11) and is often accompanied by nausea. The shape of the audiogram is often flat or rising, but any configuration is possible. It is sometimes possible to arrest (and possibly even reverse) the progression of early-onset luetic hearing loss with high doses of penicillin and steroids. Toxoplasmosis is a parasitic infection caused by the protozoan Toxoplasma gondii, and is often contracted from contaminated raw meats and eggs, as well as from contact with cat feces. The disease is transmitted to the developing fetus via the placenta, often from a mother who does not have any symptoms herself. The risk of adverse effects in the infant is great for infections incurred during the first trimester but small during late pregnancy. The incidence of toxoplasmosis is roughly 1.1 in 1000 births. Congenital toxoplasmosis causes a variety of disorders, including central nervous system disorders (e.g., microcephaly, hydrocephaly, intracranial calcifications, and intellectual disability [intellectual developmental disorder]3), chorioretinitis (inflammation of the choroid and retina) and other eye disorders, and bilateral sensorineural hearing loss that may be moderate to severe and progressive. Hearing losses have been reported in 14 to 26% of children with congenital toxoplasmosis. Optimistically, Stein and Boyer (1994) found no hearing losses in 58 children treated with antiparasitic and sulfonamide drugs for 12 months beginning when they were less than 2.5 months old, although long term follow-up studies of these children are still needed. Rubella (German measles) is a viral disease transmitted from the mother to the fetus via the placenta. Epidemics in the United States during the 1960s have probably made it the most infamous viral cause of congenital hearing impairment, but the rubella vaccine has reduced the number of congenital rubella cases to ~ 50 per year (Strasnick & Jacobson 1995). Babies affected by congenital rubella may have heart disorders, kidney disorders, intellectual developmental disorder, visual defects, and hearing loss. The risk for congenital rubella is greatest if the fetus is exposed during the first trimester (roughly 50% in the first month, 20% in the second month, and 10% in the third), but the possibility of significant risks extends out to the 16th week. Sensorineural hearing loss is the most frequent sequela, with an incidence of ~ 50% when rubella is contracted during the first trimester and ~ 20% in the second and third trimesters. Congenital rubella is typically associated with bilateral sensorineural hearing losses that are severe to profound in degree, and either flat, bowl-shaped, or sloping in configuration. Cytomegalovirus (CMV) affects 2 to 3% of live births and is probably the most common viral cause of congenital hearing loss. It is a herpes-type virus often contracted by sexual contact, and is also known to be transmitted by close associations with infected children. Pregnant women can be infected by a new exposure or by the reactivation of latent viruses already present in their bodies, and are usually asymptomatic when they do have the infection. The major consequences of CMV are associated with transmission to the developing fetus; postnatal CMV infections do not seem to present any major risks for an otherwise healthy child. The severe outcomes of congenital CMV may include death, microcephaly, pneumonia, intellectual developmental disorder, liver disease, dental defects, visual lesions, and sensorineural hearing loss (Stagno 1990; Schildroth 1994; Strasnick & Jacobson 1995). About 10% of CMV infected newborns are symptomatic, with one or more manifestations such as a purple rash, jaundice, “blueberry muffin” skin discolorations, and various signs of infection. About 90% are asymptomatic, having “silent” CMV infections. Overall, Stagno (1990) reported that 92% of symptomatic newborns have one or more serious sequela (including a 30% death rate) compared with only 6% for asymptomatic infants. About 10 to 15% of the cases eventually have one or more serious complications of congenital cytomegalovirus. Cytomegalovirus causes a wide variety of sensorineural hearing losses, ranging from mild through profound, which may be bilateral or unilateral, progressive or stable. Overall, ~ 14% of children born with CMV have some degree of sensorineural hearing loss (Schildroth 1994; Grosse, Ross, & Dollard 2008). However, while Schildroth (1994) reported that ~ 88% have severe to profound impairments and 50% have at least one additional disability, Grosse et al (2008) estimated that ~ 3 to 5% of children with CMV have bilateral losses that are moderate to profound. Herpes simplex is a sexually transmitted viral disease that can be passed from an infected mother to the developing fetus either during pregnancy or during delivery (perinatally). Infected infants are affected by central nervous system problems (e.g., microcephaly and psychomotor disorders), growth deficiencies, retinal dysplasia, and moderate to severe sensorineural hearing loss in one or both ears. Maternal infections are not the only adverse influences that can cause or contribute to hearing impairments in the developing fetus. Another group of causes includes drugs, chemicals, and other agents in the maternal environment (Strasnick & Jacobson 1995). For example, congenital hearing loss can be caused by the maternal use of certain medications that may be passed to the fetus via the placenta. The most prominent group of drugs are the aminoglycoside antibiotics, such as kanamycin, gentamicin, and streptomycin, which are sometimes essential in the treatment of severe infections. Other aspects of the maternal environment that can contribute to congenital hearing impairments and related anomalies include maternal diseases such as toxemia and diabetes, nutritional deficiencies and disturbances, Rh-factor incompatibility between the mother and fetus, exposure to physical agents such as heat and radiation, and use of non-medicinal drugs and chemicals such as alcohol. Adverse factors that occur just before, during, and immediately after birth include compromise of the oxygen supply (asphyxia, anoxia, hypoxia), trauma during labor and/or delivery, hyperbilirubinemia (jaundice) leading to kernicterus, and infections. Borg (1997) reviewed 20 years of research to clarify the relationship between hearing loss and prenatal hypoxia (oxygen deficiency), ischemia (blood supply deficiency), and asphyxia (indicated by a low Apgar score). He found that the risk of permanent sensorineural hearing loss is greater for ischemia than for hypoxia; the central nervous system (CNS) is more susceptible to these kinds of insults than is the inner ear; and preterm infants are more susceptible than full-term babies. Interestingly, hearing losses were rarely caused by birth asphyxia alone, and hypoxia all by itself appeared to be associated with temporary hearing losses as opposed to permanent ones. Some elucidation about hyperbilirubinemia is desirable because not every newborn with jaundice is at risk. The normal breakdown of spent red blood cells produces bilirubin, which is detoxified by the liver and excreted. Hyperbilirubinemia is a build-up of bilirubin in the blood and is observed as jaundice. Some degree of jaundice is common among newborns, and it is successfully treated by exposing the baby to ultraviolet light (phototherapy). However, Rh-factor incompatibilities between the mother (who is Rh-negative) and the fetus (who is Rh-positive) can result in the production of antibodies in the mother’s blood, which can attack and break down red blood cells in the fetus. This disorder is called erythroblastosis fetalis or hemolytic disease of the neonate, and usually occurs in subsequent pregnancies where the fetus is Rh-positive. The concentration of bilirubin in the blood of the fetus can become high enough to cross the blood-brain barrier, causing kernicterus, or the deposit of bilirubin in the brain. Kernicterus causes brain damage because bilirubin is toxic to neural tissue. It can be widespread, but particularly affects the basal ganglia. Typical sequelae include athetoid cerebral palsy, intellectual developmental disorder, and hearing impairment. Blood transfusions (exchange transfusions) are used to avert or minimize these effects in newborns with high bilirubin levels. A variety of sensorineural losses are found in ~ 4% of babies with hemolytic disease or hyperbilirubinemia (Hyman, Keaster, Hanson, et al 1969). However, whether hearing losses in kernicterus patients come from damage to the brainstem centers (such as the cochlear nuclei) and/or to coincidental peripheral impairments seems to be an unresolved issue. Dysplasia means an abnormality in the development of an anatomical structure. These congenital anomalies can affect the outer, middle, and/or inner ear, and can occur alone or as part of syndromes. The conductive system anomalies span a range of severity, from barely noticeable cosmetic aberrations to a complete lack of development (aplasia). Outer and middle ear anomalies may occur alone or together, as well as in combination with inner ear dysplasia, especially in more severe cases. The major external ear anomalies are microtia and atresia, which often occur together. Microtia means an abnormally small pinna, but it actually refers to a wide range of auricle deformities. Type I microtias have recognizable parts, and can be more or less well formed except for their size. Pinnas that are only partially formed, generally resembling a curved or straight ridge, are type II microtias. A type III microtia is a tissue mass that does not resemble a pinna. Finally, the auricle may be completely absent, which is called agenesis (or aplasia) of the pinna, or anotia. Microtias are a major cosmetic problem, but do not cause substantial hearing losses in and of themselves. (This is not to undervalue the important auditory role of the pinna in such processes as directional hearing.) In contrast, considerable degrees of hearing loss can be produced by aural atresia, which is the absence of an external auditory meatus. If the canal opening is abnormally narrow, then the condition is called aural stenosis. Many kinds of congenital middle ear anomalies are possible, such as these: 1. The middle ear cavity and antrum may be grossly malformed, slit-like, or altogether absent. 2. The tympanic membrane may be rudimentary or absent. 3. Ossicles may be abnormally formed; for example, the malleus and incus are often conglomerated into a unit. 4. Ossicles may be missing. 5. Ossicles may be attached to the abnormally formed middle ear cavity either directly or via bony bridges. 6. Facial nerve abnormalities are also frequently encountered. Outer and middle ear anomalies produce a conductive hearing loss, provided the inner ear is not also affected. The degree of the conductive loss depends on the nature and extent of the abnormalities and is often ~ 60 dB. Surgical reconstruction of the conductive mechanism is generally possible, provided there is a serviceable cochlea behind the abnormal conductive mechanism. Depending on the nature of the abnormal anatomy, it is often possible to reduce the conductive loss to the 30 dB range, which is a considerable degree of improvement. Surgical reconstruction for the microtia provides the child with cosmetic improvements, the importance of which should not be underestimated. The congenital anomalies of the inner ear also exist along a continuum of severity from slight to complete. The worst case is the total absence of any inner ear structures, known as Michel’s aplasia. It is possible for this to occur even with a normal conductive mechanism. Incomplete anomalies and developmental failures of the bony and membranous ducts of the inner ear are known as Mondini’s dysplasia and vary widely in severity. The most common abnormality is dysplasia of the membranous ducts of the cochlea and saccule, and is called Scheibe’s aplasia. It is generally believed that Michel’s and Mondini’s anomalies are caused by autosomal dominant traits, and that Scheibe’s aplasia is transmitted by the autosomal recessive route. Alexander’s aplasia refers to congenital abnormalities of the cochlear duct, especially affecting the basal turn (high-frequency region) of the cochlea. Traumatic head injuries can cause conductive, sensorineural, and mixed hearing losses, depending on the nature and extent of the injuries. Head trauma causes a conductive hearing loss by such mechanisms as injuries to the eardrum and/or ossicles, accumulations of blood and debris in the ear canal and middle ear, and temporal bone fractures. Traumatic sensorineural losses are typically caused by concussion to the inner ear and fractures of the temporal bone directly injuring the inner ear structures. Temporal bone fractures are classified according to whether the fracture line is in the same direction as the axis of the petrous pyramid (longitudinal) or across it (transverse). The relationships of these fractures to the structures of the ear are shown in Fig. 6.2. Longitudinal fractures go through the ear canal and middle ear and usually bypass the inner ear structures, so that they are most likely to produce conductive losses. About 80% of temporal bone fractures are longitudinal. In addition, concussion to the cochlea can result in a sensorineural loss (generally high-frequency) even though the inner ear structures are not directly involved in the longitudinal fracture. In contrast, transverse fractures cut through the inner ear, often resulting in profound sensorineural hearing loss and vertigo. Facial nerve paralysis occurs in ~ 50% of patients with transverse fractures. Mixed fractures are also common. Cerumen is a waxy substance that is supposed to be in the ear canal, where it serves lubricating and cleansing functions and also helps to protect the ear from bacteria, fungi, and insects. The cerumen is produced by glands in the cartilaginous portion of the ear canal and migrates out over time. Small amounts of cerumen often accumulate in normal ears, typically seen as yellow to brown globs that do not obstruct the ear canal. Impacted cerumen is an accumulation of wax in the ear canal that interferes with the flow of sound to the eardrum. Impacted cerumen occurs naturally in many patients who produce excessive amounts of cerumen, which builds up over time. It is also the fate of many Q-tip-wielding patients who inadvertently pack cerumen farther back into the canal (and frequently against the eardrum) in an ironic attempt to clean their ears. Fig. 6.2 Schematic representations of (a) longitudinal and (b) transverse fractures of the temporal bone and how they relate to ear structures. (Adapted from Swartz and Harnsberger [1998], with permission.) The treatment for excessive cerumen is to remove it. Cerumen management procedures (e.g., Ballachanda & Peers 1992; Roeser & Wilson 2000; Roland, Smith, Schwartz, et al 2008) are undertaken only after an otoscopic examination and a complete history are taken. Cerumen removal may be preceded by the use of a wax softening product, and is accomplished by one technique or a combination of techniques including irrigation of the ear with water, removal with a blunt curette designed for this purpose, and suction. Cerumen management is within the scope of audiological practice (e.g., ASHA 2004, 2006), with ~ 69% of audiologists overall (and ~ 87% of private practitioners) providing this service as of 2011 (Johnson, Danhauer, Rice, & Fisher 2013). A seemingly endless assortment of foreign bodies can get into the ear. Some of these may get pushed into the ear where they can produce traumatic injuries to the canal wall, tympanic membrane, and middle ear structures. Others get stuck in the canal. Others do both. Foreign bodies that get stuck in the canal may be inorganic or organic, including live insects, and can produce the same effects as impacted cerumen plus traumatic injuries and/or infection depending on the nature of the object. Foreign objects in the ear should be removed by an otologist. The outer ear can be the site of cysts as well as benign and malignant tumors that should be addressed medically. Hearing becomes a factor if the ear canal is obstructed. Exostoses are the most common tumors of the ear canals, and are regularly encountered audiologically. These are benign, skin-covered bony growths from the canal walls that are smooth and rounded. They are usually found bilaterally. The development of exostoses is encouraged by repeated encounters with cold water and are common among swimmers. It is uncommon to find exostoses that will fully occlude the external auditory meatus by themselves, but their presence makes it easier for the canal to be blocked by cerumen or debris in the canal. External otitis (otitis externa) is a diffuse infection of the external auditory meatus usually caused by pseudomonas (and less often by staphylococcus) bacteria. It is occasionally known as “swimmer’s ear” because of its association with swimming in inadequately maintained pools. External otitis produces a considerable amount of pain, as well as edema (swelling), discharge, itching, and a conductive hearing loss if the canal is occluded by the swelling and debris. It is treated with antibiotic drops or creams that are frequently mixed with hydrocortisone to help reduce the edema. When there is considerable edema a gauze wick may be impregnated with the antibiotic/cortisone cream and gently pushed into the swollen ear canal to act as a medication applicator. Otitis externa can develop into an aggressive and life-threatening form of the infection called necrotizing (malignant) external otitis in diabetics and other susceptible patients, and requires extensive antibiotic and other medical treatment. The furuncle is an example of a more circumscribed outer ear infection. It is a staphylococcus infection of a hair follicle in the cartilaginous portion of the ear canal. Furuncles are treated with oral antibiotics, the placement of an alcohol-soaked wick in the ear canal until it spontaneously opens and drains, and/or incision and drainage of the furuncle under local anesthesia. Bullus myringitis is a very painful viral infection seen as inflamed, fluid-filled blebs or blister-like sacs on the tympanic membrane and nearby ear canal walls. It often occurs in association with upper respiratory infections as well as with otitis media. Very little hearing loss is associated with this condition even though it does impair the mobility of the tympanic membrane to some extent. Bullus myringitis is usually self-limiting, and a thin fluid (perhaps with blood) is discharged when the blebs rupture. Antibiotics are often used to avoid secondary infections. Tympanosclerosis refers to a variety of tissue changes that occur as the result of repeated middle ear infections. It is often seen as chalky white calcium plaques and scar tissue on the pars tensa of the eardrum, which has little if any effect on hearing sensitivity. Tympanosclerosis also affects the other structures of the middle ear, and can affect hearing if the calcification or other changes impair the mobility of the ossicular chain. Perforations of the tympanic membrane can be caused by ear infections and various kinds of traumatic insults. Typical traumatic causes include (1) punctures; (2) chemical injuries; and (3) forceful pressure changes due to an explosion, a slap to the ear, intense sound (acoustic trauma), and sudden changes in air pressure while flying or in water pressure while diving (barotrauma). As illustrated in Fig. 6.3, the opening is described as a central, marginal, or attic perforation, according to its location on the eardrum. Attic perforations involve the pars flaccida and are often associated with cholesteatoma, as discussed below. Eardrum perforations produce conductive hearing losses because they (1) reduce the size of the drum’s vibrating surface area, which reduces the eardrum-to-oval-window area advantage; (2) impair the coupling of the eardrum to the ossicular chain so that signal transmission is reduced; and (3) interfere with the proper phase relationship at the oval and round windows by allowing sound to impact upon the round window. In general, the magnitude of the conductive hearing loss gets worse as the size of the perforation increases. Many tympanic membrane perforations heal spontaneously. Healed perforations are associated with the development of scar tissue and thin areas that do not have a fibrous middle layer, which are sometimes called monomeric membranes. Larger perforations and those associated with chronic middle ear infections generally do not heal on their own, and require a surgical repair of the eardrum called a myringoplasty. Inflammations of the middle ear are called otitis media, and constitute the most common cause of conductive hearing losses. Otitis media affects people of all ages, but the incidence among children is particularly high. For example, incidence figures have been reported at over 50% by age 1 and 60% by age 2, ~ 33% for at least three episodes by age 3, roughly 80% for children in day care centers, and ~ 90% by the time a child reaches school age (e.g., Denny 1984; Teele, Klein, & Rosner 1984; Prellner, Kalm, & Harsten 1992; AAP 2004). The implications of otitis media in children go beyond the medical ramifications of the pathology and the direct interference with communication caused by the conductive hearing loss. Young children with recurrent otitis media are subjected to frequent and sustained periods of conductive hearing impairment during critical learning periods. For example, Gravel and Wallace (2000) found that during years 1 through 3, children who had otitis media with effusion (OME) had consistently poorer hearing thresholds than their OME-free counterparts. For this reason, it is not surprising that recurrent otitis media in young children has been associated with deficits affecting auditory processing, language development, cognitive and academic skills (e.g., Jerger, Jerger, Alford, & Abrams 1983; Friel-Patti & Finitzo 1990; Menjuk 1992; Gravel & Wallace 1992; Brown 1994; Downs 1995; Schwartz, Mody, & Petinou 1997; Mody, Schwartz, Gravel, & Ruben 1999). It is interesting to note here that the temporal characteristics of auditory cortex activity were changed by 1 to 2 weeks of experimentally induced conductive hearing losses in developing gerbils (Xu, Kotak, & Sanes 2007). Fig. 6.3 Examples of central, marginal, and attic perforations of the tympanic membrane. (Adapted from Hughes [1985], with permission.) Otitis media can take several forms depending on such factors as the development and time course of the disease, whether a bacterial infection is present, the nature of the fluid in the middle ear space, and the kinds of complications that exist. One way to address these issues is to trace the course that the disease might follow. A properly functioning Eustachian tube provides the middle ear with ventilation and drainage, and maintains the same amount of pressure on both sides of the tympanic membrane. Recall from Chapter 2 that the Eustachian tube is normally closed, and that it is opened by the tensor palatini muscle during activities such as chewing and yawning; and we shall soon see that problems with this process can lead to an accumulation of fluid in the middle ear and the possible development of more serious disease. It is interesting to note in this context that middle ear fluid was found to be significantly less common among 2- to 6-year olds who regularly chew gum compared with those who do not (Kouwen & DeJonckere 2007). Eustachian tube dysfunction exists when the tube does not open properly, is not patent, or is blocked in one way or another. The tube might be blocked by edema (swelling) and/or fluid caused by an upper respiratory infection, sinusitis, inflamed adenoids, or allergies; obstruction by hypertrophic (enlarged) adenoids; or obstruction or encroachment by a nasopharyngeal tumor or other growth. The Eustachian tube may also fail to open appropriately (e.g., upon swallowing or yawning) due to structural or functional abnormalities affecting the tensor palatini muscle. This problem is common among cleft palate patients. Unlike the adult Eustachian tube, which tilts downward by ~ 45°, infants and young children have almost horizontal Eustachian tubes (Fig. 6.4), which are also relatively shorter and wider, in addition to which their tensor palatini muscles operate less efficiently. These factors increase the chances of Eustachian tube dysfunction and middle ear disease among infants and young children with growth and development by age 6 or 7 (Rovers, Schilder, Zielhuis, & Rosenfeld 2004). Eustachian tube dysfunction prevents the middle ear from being ventilated, causing the air within the middle ear cavity to become stagnant. Part of the stagnant air is then absorbed by the tissues of the middle ear. As a result, the air pressure will be lower inside the middle ear than it is outside in the surrounding atmosphere. This situation is called negative pressure in the middle ear.4 This pressure difference on the two sides of the tympanic membrane (higher outside, lower inside) causes it to be drawn inward, or retracted. The same nonpatency of the Eustachian tube that caused the air to be trapped within the middle ear cavity also prevents ventilation of the middle ear that would re-equalize the pressure and “unclog” the ear. If this problem is caused by an abrupt pressure change, it is called aerotitis or barotrauma. Aerotitis is usually caused by the abrupt air pressure increase of an airplane descent, which causes the tympanic membrane to retract and prevents the Eustachian tube from opening. 4 The nonpatent Eustachian tube explanation appears to account for negative pressures up to only about -100 daPa (Cantekin et al, 1980; Yee & Cantekin, 1986), so that other mechanisms may also be involved in creating abnormally negative middle ear pressures. Fig. 6.4 The Eustachian tube slopes downward in adults but is closer to being horizontal in infants and young children. (Adapted from Pulec and Horwitz [1973], with permission.) Fluid might accumulate in the middle ear, typically because it was drawn from the middle ear tissues by negative pressure caused by Eustachian tube dysfunction or allergies, or as an inflammatory response after an acute infection (e.g., Gates, Klein, Lim, et al 2002; AAP 2004). The presence of this fluid (effusion) in the middle ear in the absence of an acute infection is called otitis media with effusion (OME) (e.g., Stool, Berg, Berman, et al 1994; AAP 2004). Not surprisingly, otitis media is associated with conductive hearing loss. Since the effusion contains serous fluid, which is a watery and clear transudate that is free of cells and other materials, the condition is often called serous otitis media. The term middle ear effusion (MEE) is often used here as well, but different kinds of fluids may also accumulate in the middle ear. Continuation of the disease process may involve mucoid fluid from secretory (goblet) cells in the middle ear mucosa. This cloudy exudate is thicker and more viscous than serous fluid, containing white blood cells and other cellular material. At this stage the condition might be described as secretory otitis media, and the term mucoid otitis media may eventually be applied with the continued thickening of the effusion. Because serous fluid is absorbed by the middle ear mucosa over time, it is possible for the effusion can become progressively thicker and more viscous, eventually leading to adhesive otitis media or “glue ear,” preventing motion of the ossicles. Blood in the middle ear is called hemotympanum. Middle ear fluid can sometimes be detected otoscopically by seeing a fluid line or meniscus or by the presence of bubbles. It is often not possible to distinguish between the presence versus absence of effusion if the tympanum is completely filled with serous fluid. The diagnosis of middle ear effusion involves the use of a pneumatic (Siegel) otoscope (AAP 2004), which has a rubber bulb and tube that allows the examiner to change the air pressure against the eardrum (by squeezing the bulb). Middle ear fluid is indicated if the air pressure fails to cause observable eardrum movement. So far we have been assuming that the patient has a middle ear effusion that is nonsuppurative or nonpurulent, which means that it is free of bacterial infection. However, infections can readily develop once microbes gain access to the middle ear, usually from the nasopharynx via the Eustachian tube. Otitis media becomes suppurative or purulent when bacteria invade the middle ear system. Acute (purulent or suppurative) otitis media typically occurs during or soon after an upper respiratory infection. The most common bacteria responsible for acute suppurative or purulent otitis media are Streptococcus pneumoniae, Haemophilus influenzae, and Branhamella catarrhalis. The typical course of events often begins with hyperemia (engorgement of the tissues with blood), often seen as reddening on the tympanic membrane, accompanied by otalgia (ear pain) and possibly fever. This may be followed by exudation, in which the middle ear space is filled with fluid containing white blood cells, red blood cells, mucus, and other materials. The eardrum is red and thickened so that landmarks cannot be seen, and it bulges due to pressure from the exudate in the middle ear. This comes with increased pain, fever, and a conductive hearing loss. Building pressure can cause the eardrum to rupture, resulting in the drainage of pus and other materials from the infected middle ear. Such a purulent discharge from the ear is called otorrhea. It is often preferable for the drainage to be effected by a small surgical incision of the eardrum (called a myringotomy, which is discussed below). Drainage relieves the pressure and pain, and there is often spontaneous healing of the tympanic membrane. Ongoing or repeated episodes of acute otitis media are called chronic otitis media, which is often associated with a perforated tympanic membrane. However, ongoing ear pathology can occur even without a perforation. Hence, the distinction between acute and chronic otitis media may also be made on the basis of how long the disease lasts. For example, a case of otitis media might be called acute if it lasts up to 3 weeks, chronic if it lasts more than 12 weeks, and subacute for intermediate durations. Some cases of chronic otitis media with intact eardrums are not detected clinically and may even be undetectable, and have been termed silent otitis media (Paparella, Goycoolea, Bassiouni, & Koutroupas 1986). As one might expect, patients who experience repeated episodes over time are said to have recurrent otitis media. Otitis media can result in quite a few sequelae and complications of major medical significance, some of which can have life-threatening consequences. Cholesteatoma is a common problem, and is discussed below. Facial paralysis can result if the infection erodes the Fallopian canal and affects the seventh cranial nerve. Spread of the infection to the inner ear results in labyrinthitis. Recall that the tympanum communicates with the mastoid antrum and air cell system, so that otitis media can spread to infect the mastoid, producing mastoiditis. Mastoiditis can lead to the further spread of the infection to the central nervous system, as well as to other sites. Conductive hearing loss is certainly the effect with which we are most directly concerned. Any amount of conductive loss can occur with otitis media, and fluctuation is common. These audiograms tend to be reasonably flat, but it is not uncommon to find losses that are poorer in the low frequencies, and others that are somewhat tent-shaped (Fig. 6.5). Otitis media can also cause a sensorineural loss, particularly in the high frequencies. The sensorineural component may be caused by the transmission of toxins to the perilymph via the round window. However, conductive lesions can affect bone-conduction thresholds, which might account for some of the sensorineural components seen in these patients, at least in part (see Chapter 5, and the section on otosclerosis in this chapter). Otitis media can lead to a continuing conductive hearing loss by causing such conditions as (1) perforated eardrums; (2) adhesions, tympanosclerosis, and glue ear, which restrict or prevent motion of the ossicles; and (3) ossicular chain discontinuities that occur when the infective process causes part of the chain to be eroded away. A cholesteatoma is a cyst made of layers of keratin producing squamous epithelium filled with an accumulation of keratin and cellular debris. It is also known as a keratoma or an epidermoid inclusion cyst. Even though keratoma is the less popular term, it is actually more appropriate because cholesteatomas contain considerable amounts of keratin but little if any cholesterol. Cholesteatomas are usually associated with chronic otitis media, and with perforated and retracted tympanic membranes. They also occur spontaneously in a small percentage of patients and can develop in the middle ear, eardrum, or petrous portion, and even intracranially. The most common sites for cholesteatoma formation are pars flaccida and marginal perforations. Let us consider the development of an attic cholesteatoma, which is a very common type. The cholesteatoma might begin as a small retracted pouch (retraction pocket) in the pars flaccida, often encouraged by ongoing negative pressure in the middle ear. The sac retains keratin and debris. Inflammation of the sac causes it to swell and expand. The cholesteatoma may lie dormant for some time, or it may grow slowly or quickly. The invasion route begins in the epitympanic recess, or attic, as shown in Fig. 6.6. Notice in the figure that a cholesteatoma that appears tiny when viewed from the outside can actually be quite large. The expanding cholesteatoma destructively encroaches upon the middle ear space and structures. As it continues to grow, the cholesteatoma has the potential to invade other locations, such as the mastoid, labyrinth, and cranium. This aggressive capability makes the cholesteatoma a potentially life-threatening lesion that must be removed surgically. Fig. 6.5 (a–c) Audiograms from three patients with otitis media.
 The Case History
The Case History
 Conductive, Sensorineural, and Mixed Impairments
Conductive, Sensorineural, and Mixed Impairments
 Tinnitus and Hyperacusis
Tinnitus and Hyperacusis
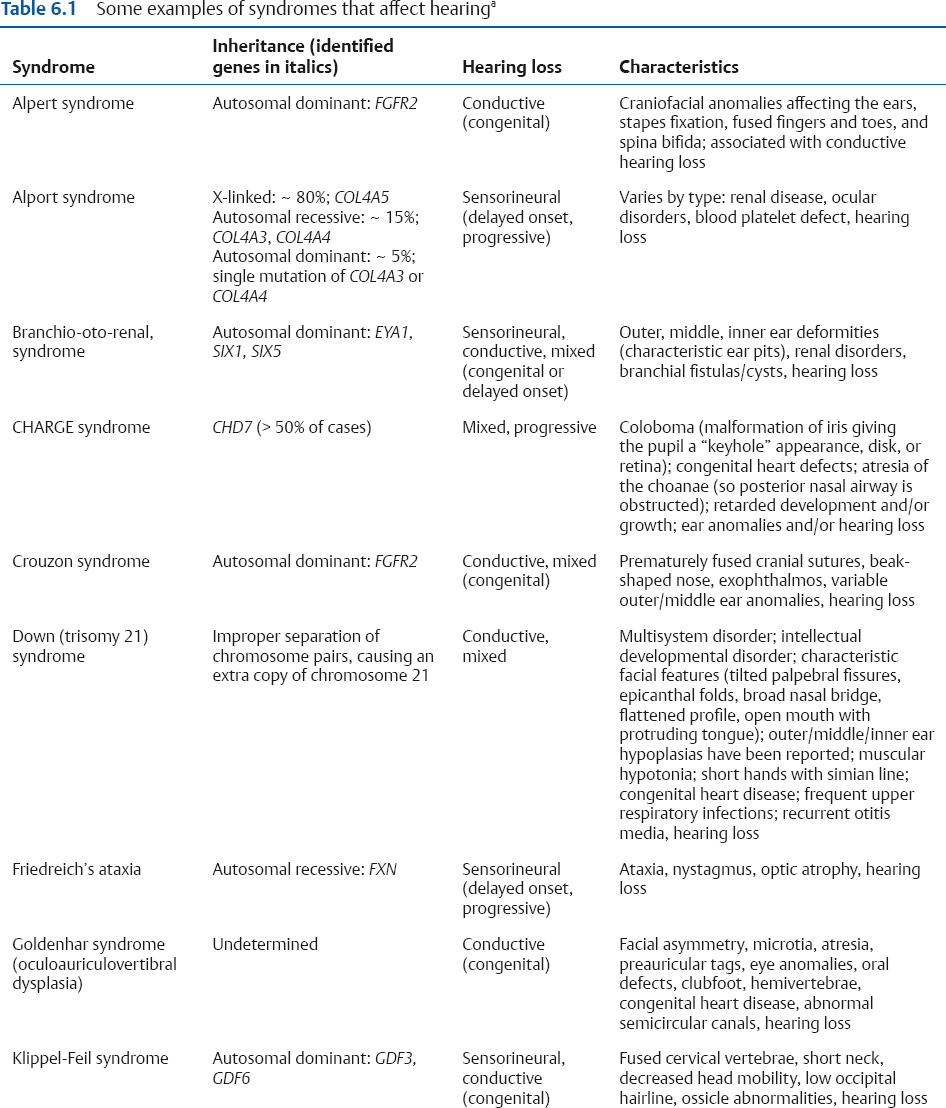
 Congenital and Hereditary Disorders
Congenital and Hereditary Disorders
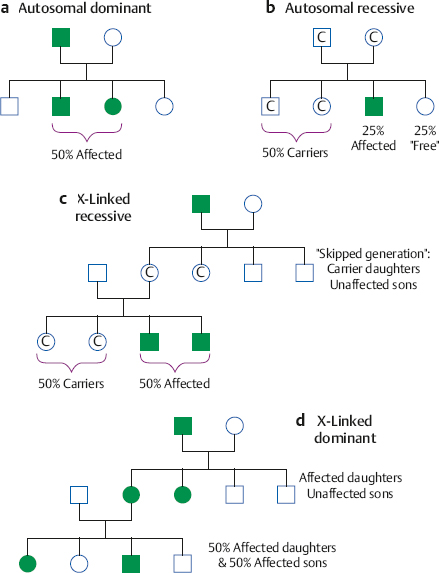
Genetic Influences
Nonsyndromic Hearing Loss
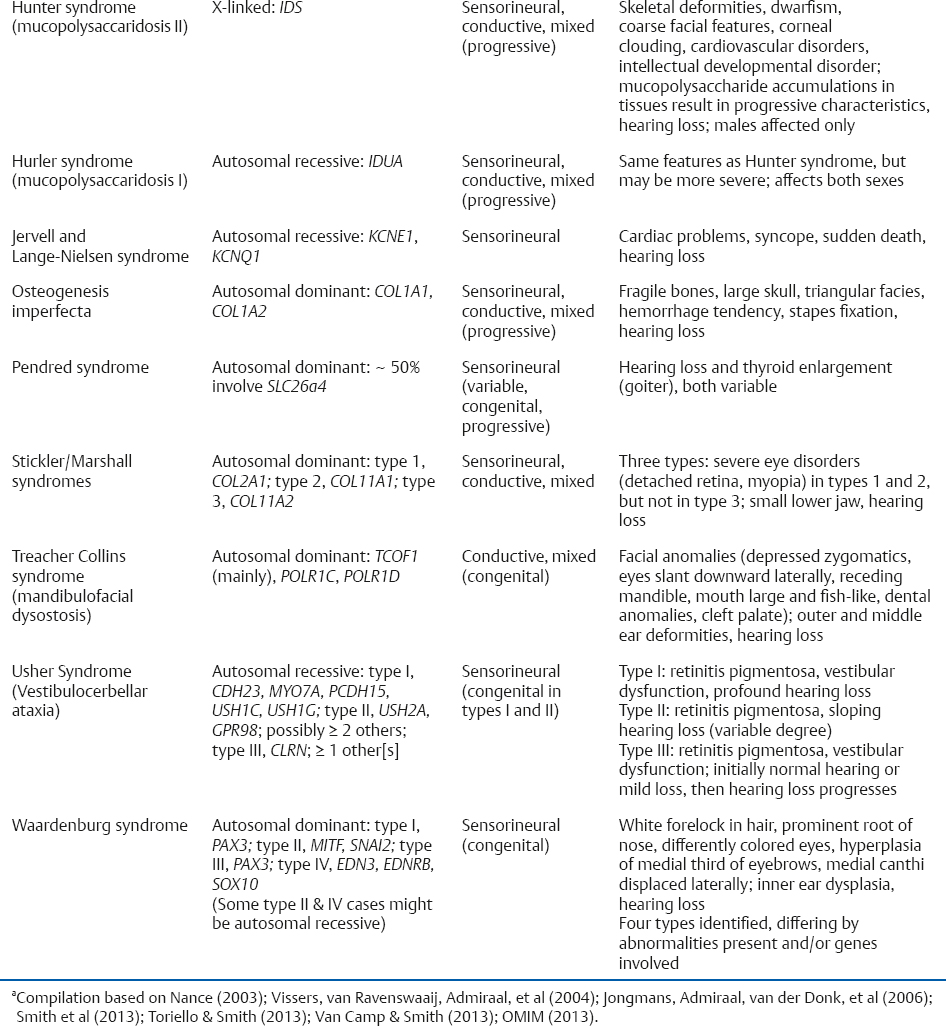
Syndromic Hearing Loss
Maternal Infections
Other Influences in the Maternal Environment
Congenital Anomalies of the Ear
Outer and Middle Ear
Inner Ear
 Acquired Disorders
Acquired Disorders
Head Trauma
 Outer Ear Disorders
Outer Ear Disorders
Impacted Cerumen
Foreign Bodies
Growths and Tumors
Infections
 Middle Ear Disorders
Middle Ear Disorders
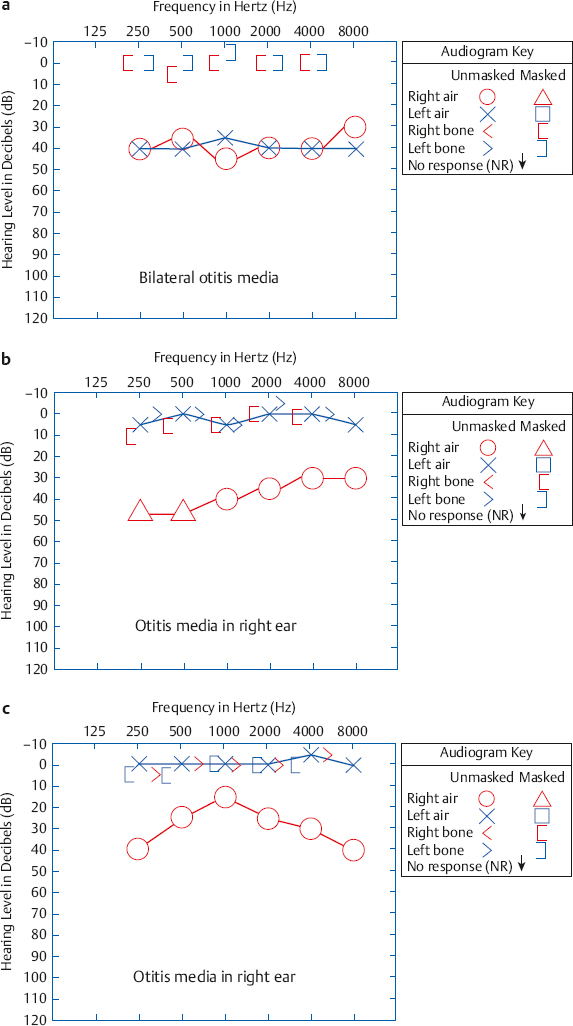
Otitis Media and Associated Pathologies
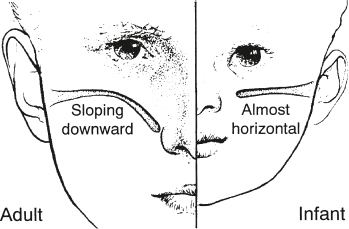
Eustachian Tube Dysfunction
Otitis Media with Effusion
Acute Otitis Media
Complications and Sequelae of Otitis Media
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


