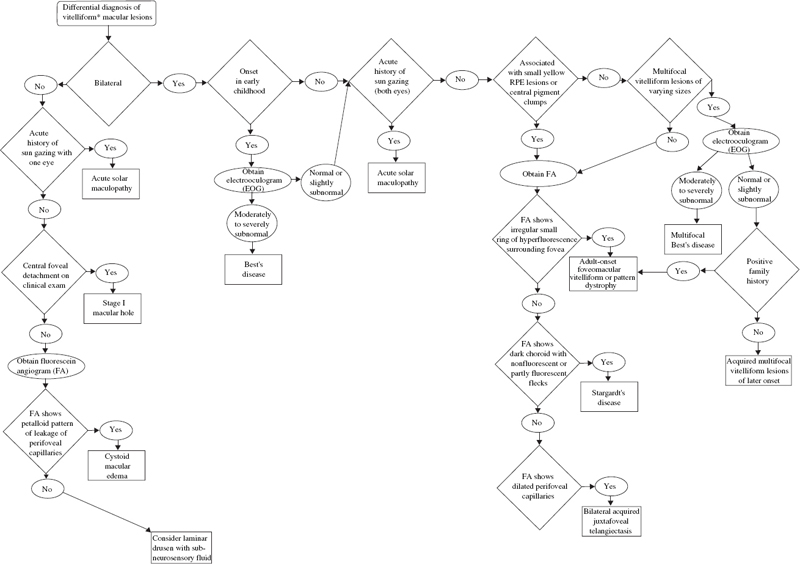
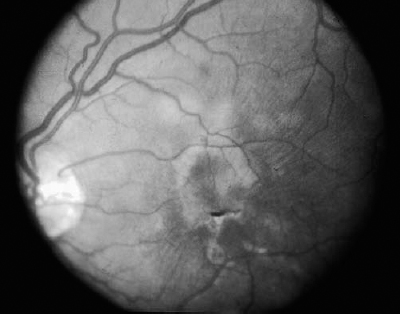
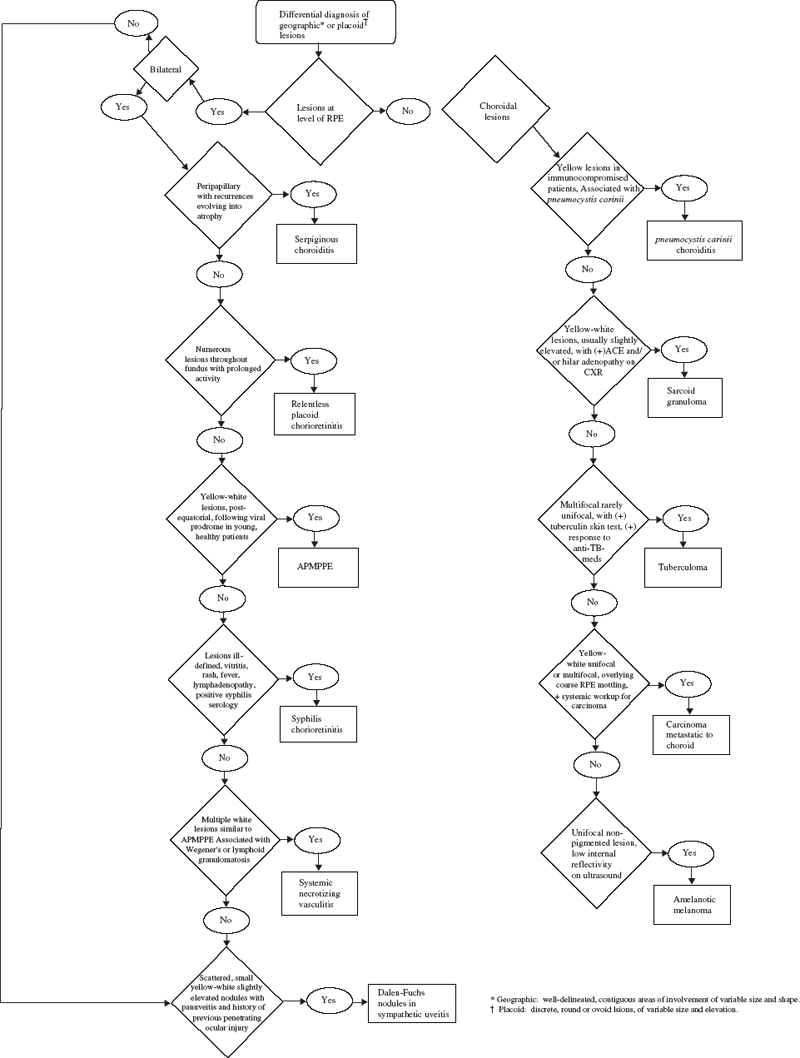
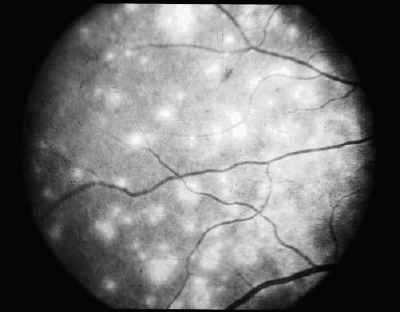
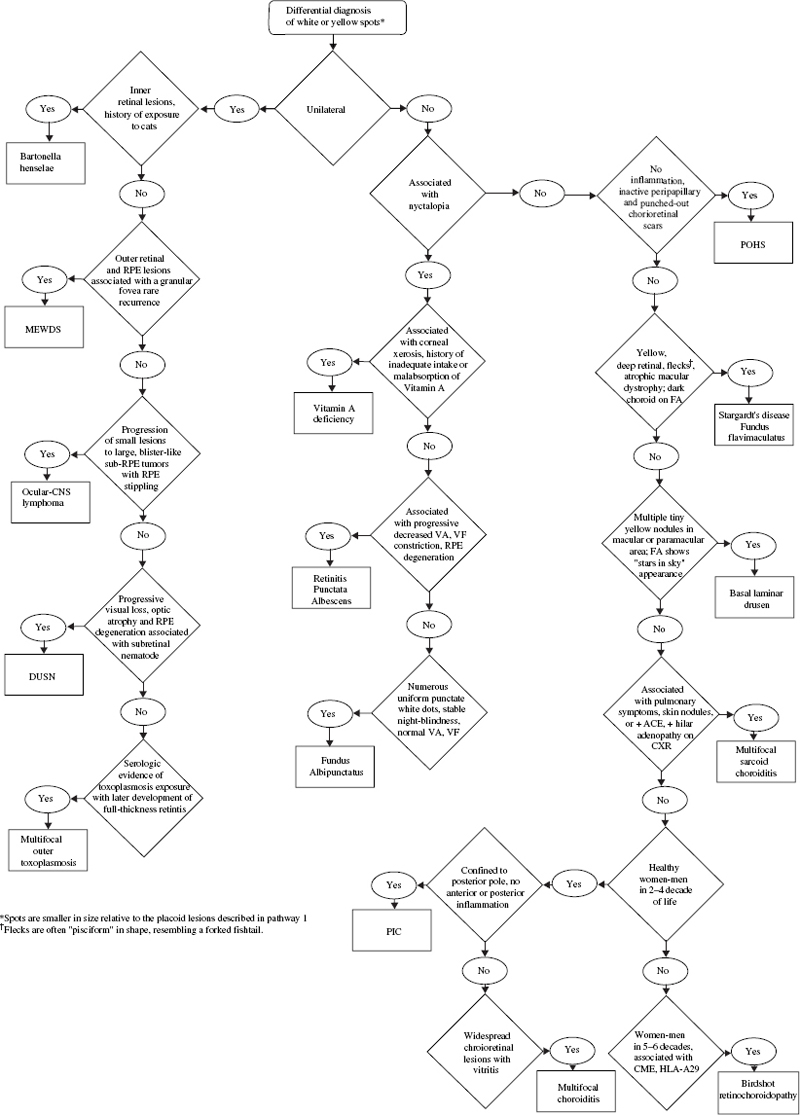
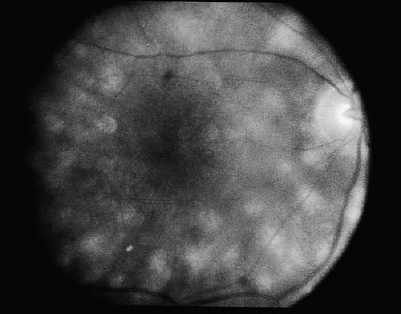
SECTION II The typical patient is a healthy young adult in the second or third decade of life. Men and women are affected equally.1–5 The patient develops rapid, painless loss of vision in one or, more commonly, both eyes.1–5 Many patients report an antecedent viral illness.2,4,6 Symptoms of meningismus, headaches, and transient hearing loss may develop.7,8 Cerebral vasculitis, rarely fatal, with spinal fluid pleocytosis and elevated protein can occur.9–13 Acute nephritis with hematuria has been reported.14,15 Multiple flat, creamy white, placoid lesions are seen at the level of the retinal pigment epithelium (RPE) and outer retina (Fig. 6–1). They are typically bilateral and found in the postequatorial retina.1–5 Inflammatory cells may be seen in the vitreous or anterior chamber. Associated episcleritis,16 disc edema,16 and perivenous exudation1 have been reported. Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) should be distinguished from other conditions presenting with geographic or placoid lesions (Fig. 6–2). Early hypofluorescence with late staining of the lesions is seen on fluorescein angiography.1 Hypofluorescence of lesions is also seen on indocyanine green (ICG) angiography.17 Within 1 to 2 weeks following onset of symptoms, the lesions begin to fade and evolve into areas of RPE atrophy and clumping. The visual prognosis is excellent: Most patients recover 20/30 or better vision. The vision typically improves rapidly within the first 2 to 3 weeks, although in a few patients visual recovery may take up to 6 months. Recurrences, although rare, do occur,4,18,19 and choroidal neovascularization has been reported.20 Residual paracentral scotomata may persist in some patients. The natural history of the disease is favorable without treatment. There is no evidence that corticosteroids speed visual recovery or improve visual outcome, although a short course may be justified in cases where the fovea is involved and visual acuity is poor. Multiple evanescent white-dot syndrome (MEWDS) typically affects young, healthy women.21–23 Patients frequently present with blurred vision, shimmering photopsias (especially in the temporal field), and paracentral scotomata.21–23 Patients frequently report a flulike illness prior to the onset of visual symptoms.21 Elevated serum immunoglobulin M (IgM) and immunoglobulin G (IgG) values have been detected in a patient with MEWDS.24 Cases of MEWDS following varicella infection25 and after hepatitis B vaccination26 have been reported. FIGURE 6–1. Acute posterior multifocal placoid pigment epitheliopathy (APMPPE). Creamy white placoid lesions at the level of the RPE are seen in differing stages of evolution. Pigment clumping is associated with the older, central lesion, whereas newer, smaller lesions are seen at its periphery. Typically, MEWDS is unilateral, although bilateral MEWDS, which may be either simultaneous or sequential, has been reported.22,27–29 This disorder is characterized by multiple small, gray-white spots found in the perimacular area at the level of the RPE and outer retina (Fig. 6–3) and may be associated with vitreous cells. A granular foveal appearance can help to distinguish MEWDS from other retinal “white-dot” disorders (Fig. 6–4). Optic disc edema and retinal vascular sheathing may also be present. Early hyperfluorescence with late staining of the lesions, often in a wreath-shaped pattern, is seen on fluorescein angiography.21,23 Leakage from disc capillaries also may be seen. ICG angiography reveals multiple hypofluorescent lesions, some corresponding to the white dots.30 An enlarged blind spot may be seen on visual field testing.31,32 Electroretinography may demonstrate profoundly decreased a-wave and b-wave amplitudes acutely, with normalization of the electroretinogram (ERG) during the recovery phase.33 Visual acuity returns to baseline, the white dots fade, and disc edema resolves over the course of several weeks. It may take months before photopsias and scotomata fully resolve. A chronic form of bilateral MEWDS with multiple recurrences has been reported.27 Usually, MEWDS is a self-limited disease with an excellent visual prognosis, and no treatment is indicated. Women are affected more commonly than men (60% versus 40%), typically in the fifth and sixth decades of life.34,35 The age at presentation is older than that of other “white-dot” disorders, such as multifocal choroiditis (Fig. 6–4). Patients note floaters, blurred vision, and photopsias initially, with nyctalopia and color vision deficits later in the course of the disease.34,35 Multiple ill-defined, yellowish, deep retinal or choroidal lesions posterior to the equator are associated with vitreous inflammation, optic disc edema, and cystoid macular edema34–36 (Fig. 6–5). Many of the lesions show no change in background choroidal fluorescence, although some demonstrate early hypofluorescence with late hyperfluorescence. Perivascular leakage, optic disc staining, and cystoid macular edema are seen.34–36 ICG angiography may reveal hypofluorescent spots. The association of birdshot with human leukocyte antigen (HLA)-A29 (90%) is the strongest of all HLA-associated diseases.37,38 Electroretinography is typically abnormal.36,39 Electrooculography may be either normal or subnormal.35,39 FIGURE 6–2. Differential diagnosis of geographic or placoid lesions. RPE, retinal pigment epithelium; APMPPE, acute posterior multifocal placoid pigment epitheliopathy; CXR, chest x-ray; ACE, angiotensin convecting enzyme. FIGURE 6–3. Multiple evanescent white-dot syndrome (MEWDS). Multiple gray-white spots at the level of the retinal pigment epithelium and outer retina are seen just nasal to the optic nerve. This is a particularly severe case of MEWDS; in other patients the lesions may be fewer and more subtle. Birdshot retinochoroidopathy is a chronic disease characterized by remissions and exacerbations. Half of affected eyes can be expected to retain a visual acuity of 20/60 or better.36 Chronic cystoid macular edema is the most common cause of visual acuity loss. Choroidal neovascularization, macular epiretinal membrane formation, macular hole, macular scar, and cataract also may be responsible for reduced visual acuity. Initial treatment consists of systemic or periocular corticosteroids. The use of cyclosporine, cyclophosphamide, azathioprine, or methotrexate may be indicated for patients with systemic or ocular steroid intolerance or for those with progressive visual loss despite steroid therapy.40 Low doses of systemic corticosteroids or immunosuppressive agents may be required for sustained suppression of intraocular inflammation. Patients typically present with symptoms in the first two decades of life. Patients affected with classic Stargardt disease demonstrate an autosomal-recessive mode of inheritance, although a few pedigrees with a condition resembling Stargardt disease have been reported to exhibit an autosomal-dominant inheritance pattern.41,42 Autosomal-recessive Stargardt disease has been mapped to the ABCR gene,43 an adenosine triphosphate (ATP)-binding transporter gene located on the short arm of chromosome 1.44 Autosomal-dominant Stargardt disease has been mapped to the short arm of chromosome 641 and the long arm of chromosome 13.42 Patients with Stargardt disease usually present with gradual bilateral visual loss during childhood or early adulthood. Patients without macular changes or with late macular involvement may not experience symptoms until midlife. Symptoms of cone dysfunction may accompany loss of central vision.45,46 Historically, the term fundus flavimaculatus has been used to describe cases of yellowish deep retinal flecks either confined to the posterior pole or extending to the midperipheral fundus, and the term Stargardt disease has referred to an atrophic macular dystrophy associated with similar flecks. Both conditions have been mapped to the ABCR gene on the short arm of chromosome 1.44 These terms are now frequently used interchangeably in the literature and are considered to represent different expressions of Stargardt disease.45,46 The appearance may correlate with the specific mutation in the ABCR gene, but intrafamilial variable expression suggests modifying factors. Initially, the fundus may appear relatively normal, or it may exhibit a vermillion color because of lipofuscin storage in the RPE that blocks choroidal detail. Loss of the foveolar reflex may be the first ophthalmoscopic sign. An oval or bull’s-eye area of atrophic macular change described as “beaten bronze” commonly develops. Flecks may not be present early in life and then may be observed on subsequent follow-up examinations.45,47,48 The flecks commonly surround the area of macular atrophy and may extend into the midperiphery (Fig. 6–6). Some patients develop diffuse atrophy of the RPE and narrowing of the retinal vessels in addition to the changes already noted.45 Choroidal fluorescence is often obscured on fluorescein angiography by diffuse lipofuscin storage in the RPE, resulting in a “dark” or “silent” choroid that allows retinal capillaries to be more clearly visible. The presence of a dark choroid is found in most patients and may be helpful in distinguishing Stargardt disease from other disorders (Fig. 6–4). A dark choroid is not absolutely necessary for the diagnosis of Stargardt disease49 and in early life may not be present, subsequently developing along with other manifestations of the disorder.50 Hyperfluorescent window defects are seen in areas of macular atrophy. Flecks may show an irregular pattern of hyperfluorescence or may appear nonfluorescent. FIGURE 6–4. Differential diagnosis of white or yellow retinal spots. CNS, central nervous system; MEWDS, multiple evanescent white-dot syndrome; DUSN, diffuse unilateral subacute neuroretinitis; POHS, presumed ocular histoplasmosis syndrome; FA, florescein angiography; RPE, retinal pigment epithelium; HLA, human leukocyte antigen; VF, vision field; VA, visual acuity; PIC, punctate inner choroidopathy; FA, fluorescein angiography; CME, cystoid macular edema. FIGURE 6–5. Birdshot retinochoroidopathy. Multiple ill-defined deep retinal or choroidal lesions are found in the macula and the post-equatorial retina. The macular details are hazy because of vitreous inflammation. Cystoid macular edema is present. Electroretinography is usually normal, although the amplitudes may be moderately decreased in long-standing cases.45,46,50 The electrooculogram (EOG) also may be subnormal.45,46,50 Color vision testing can show a mild red-green dyschromatopsia.45,46,50 Some patients exhibit a prolongation in dark adaptation.45 FIGURE 6–6. Stargardt disease. A central area of “beaten-bronze” macular atrophy with pigment clumping is present, surrounded by numerous deep retinal flecks. Once visual acuity decreases to a level of 20/40, many patients exhibit a rather rapid deterioration of vision to 20/200.49 Vision tends to stabilize at 20/200, although it may fall to finger counting in longstanding cases. Although visual loss is usually symmetric, asymmetry may exist between the two eyes. The clinical course may be influenced by the specific mutations present and perhaps by other modifying genes or environmental factors. Whereas no treatment exists for Stargardt disease at present, the avoidance of light exposure (i.e., wearing sunglasses) may be beneficial. The International Epidemiological Age-Related Maculopathy Study Group has defined age-related maculopathy (ARM), also called age-related macular degeneration, as a degenerative disorder of persons aged 50 years or older. It is characterized by the presence of soft drusen at least 63 μ in the greatest linear dimension, and areas of hyperpigmentation or hypopigmentation associated with drusen.51 The incidence and prevalence of ARM increase with advancing age. Ethnicity appears to be an important determinant of ARM. Early ARM is observed more frequently among white than among black and Hispanic persons,52–54, and the rate of visual loss from choroidal neovascularization is much greater in white populations. Certain families have been reported to manifest a predilection for the development of ARM.55–57 The respective roles of genetics or of shared environmental factors in these familial cases have yet to be elucidated. Many patients with early ARM are asymptomatic, and the diagnosis is made on routine ophthalmic examination. Patients with centrally located drusen may present with blurred vision or metamorphopsia. Difficulty with reading, especially in dim light, and decreased dark adaptation are common symptoms. Contrast sensitivity is frequently diminished, at times to a greater extent than Snellen acuity. An acute drop in visual acuity or the abrupt onset of metamorphopsia is suggestive of neovascular ARM. Nonneovascular ARM is characterized by drusen, geographic atrophy, and focal hypopigmentation or hyperpigmentation of the RPE. Drusen are focal, whitish yellow sub-RPE nodules that vary widely in size and morphology. They may be categorized as hard (small, round, and discrete) or as soft (larger and poorly defined), or calcific. Drusen typically are clustered in the macula, but they may be found anywhere in the fundus. Whereas the number and size of drusen tend to increase with time, in some cases they disappear, occasionally leaving areas of geographic atrophy in their wake. In neovascular ARM, new blood vessels grow from the choriocapillaris through damaged Bruch membrane into the sub-RPE or subretinal space. Clinical manifestations may include intraretinal, subretinal, or sub-RPE blood; fluid beneath the RPE or retina; cystoid macular edema; intraretinal or subretinal hard exudates; subretinal gray or pigmented membranes; or subretinal fibrous tissue. Well-defined, blister-like retinal pigment epithelial detachments (PEDs) are seen in ARM and may result from blood (hemorrhagic PED), fluid (serous PED), or choroidal neovascularization (fibrovascular PED) beneath the RPE, or from coalescent drusen (drusenoid PED). The appearance of drusen on fluorescein angiography is variable. Hard drusen more frequently hyperfluoresce early and fade with the background choroidal fluorescence, whereas larger, soft drusen typically do not fluoresce until the later phases of the angiogram. The cause of retinal PEDs may be elucidated by fluorescein angiography. Hemorrhagic PEDs show blocked fluorescence; serous PEDs exhibit early, well-delineated, uniform hyperfluorescence that persists into the late phase of the angiogram; fibrovascular PEDs present with stippled hyperfluorescence, which leaks in the late phase; drusenoid PEDs hyperfluoresce early or late and variably stain in the late phase of angiography. Choroidal neovascularization (CNV) is classified as either classic or occult. Classic CNV can be identified by its early, well-delineated hyperfluorescence that leaks into the later phases of the angiogram.58 Discrete vessels may be identified in the early phases. Occult CNV is characterized by two types of patterns on fluorescein angiography: (1) a fibrovascular pigment epithelial detachment, an irregular elevation of the RPE with ill-defined, stippled hyperfluorescence in the midphase of the angiogram and late leakage; or (2) late-phase leakage of an undetermined source.58 Often ICG angiography is of benefit in determining the presence of CNV or in better localizing CNV in the presence of hemorrhage or with serous PEDs, where the bright early hyperfluorescence on fluorescein angiography may obscure the CNV. Choroidal neovascularization may appear as a “hot spot,” a small, discrete area of hyperfluorescence one disc area or smaller in size; as a plaque, an area of hyperfluorescence greater than one disc area in size that may be well or poorly defined; or as a combination of the two.59 Patients with drusen and RPE mottling frequently exhibit only mild to moderate visual loss. Those who develop advanced geographic atrophy of the central macula or neovascular ARM are likely to become legally blind in that eye. Patients over the age of 65 with bilateral drusen have a 24% chance over 3 years of developing a new atrophic or neovascular lesion that results in visual loss.60 Multiple large drusen and focal RPE hyperpigmentation are high-risk features for subsequent choroidal neovascularization.61 Patients with CNV in one eye have a reported risk of 7 to 10% per year of developing CNV in the fellow eye;62,63 this risk increases to 60% over 5 years if both large drusen and RPE hyperpigmentation are present.63 There is currently no proven treatment for nonneovascular ARM. Dietary intake of green, leafy vegetables containing the carotenoids lutein and zeaxanthin was found to be associated with a decreased risk of severe ARM.64 The Age-related Eye Disease Study (AREDS) (see Chapter 21) demonstrated the efficacy or evaluated the side effects of micronutrient supplementation. A strong positive association between smoking and both wet and dry ARM has been demonstrated in large, prospective studies,65,66 and smoking cessation should be strongly encouraged. Neovascular ARM with extrafoveal or juxtafoveal CNV should be treated according to the Macular Photocoagulation Study (MPS) guidelines.67,68 For subfoveal CNV that is predominantly classic, photodynamic therapy (PDT) with verteporfin has been shown to reduce the risk of visual loss.69 Verteporfin therapy also has been demonstrated to reduce the risk of moderate and severe vision loss significantly in subfoveal CNV that is occult with no classic component.70 Patients of all racial backgrounds may be affected by basal laminar drusen, which present in early adulthood to middle age.71,72 No pattern of heredity has been established. Patients are characteristically asymptomatic unless a vitelliform serous exudative detachment develops; this is when patients typically present with metamorphopsia and a moderate degree of visual loss.71,73 The vast majority of patients with this condition have no associated systemic condition; however, basal laminar drusen have been reported to occur in patients with type II membranoproliferative glomerulonephritis.74,75 These patients may develop choroidal neovascularization at an early age.76 A myriad of tiny, discrete, round, yellow nodules that may be scattered randomly or grouped in clusters in the macular or paramacular area are seen on fundus examination. These drusen are more apparent angiographically than clinically.71,72 Some patients, usually middle-aged or older, may develop yellow serous exudative detachments of the macula in one or both eyes.71–73 These detachments may resemble the vitelliform lesions seen in Best disease or with pattern dystrophy (Fig. 6–7). The subretinal fluid typically resolves spontaneously over the course of several months. Although some patients regain excellent visual acuity, in others geographic atrophy and poor visual acuity result following resolution of the fluid.72 Hundreds of drusen fluoresce brightly in the early phases of the fluorescein angiogram, creating a “stars in the sky” appearance.71,72 In serous detachments, the yellow subretinal fluid typically blocks fluorescence early in the course of the angiogram, with late fluorescein filling of the detachment.71–73 Both ERG and EOG are normal in patients with basal laminar drusen.72 Many patients retain excellent visual acuity. Those patients who develop geographic atrophy following resolution of serous detachments may have significant loss of vision. Choroidal neovascularization may develop, although probably less frequently than in those patients with ARM.71 There is no known treatment for basal laminar drusen or for the vitelliform serous detachments that may occur. In the event that choroidal neovascularization develops, the treatments for CNV apply. Best’s disease is inherited in an autosomal-dominant fashion with variable penetrance and expressivity. Some persons who carry the defective gene have a normal fundus and visual acuity, although their EOGs exhibit the same decrement as affected patients.77 Men and women are equally affected. The gene for Best disease has been mapped to chromosome 11q13.78 Visual acuity is initially normal, and the early lesions may be found on routine ophthalmoscopy. Decreased visual acuity is the main symptom and most frequently presents in later childhood or young adulthood. Best’s disease evolves through a series of stages.79,80 In the previtelliform stage, the fundus appears normal or may exhibit a small yellow dot centrally. In most cases, the typical vitelliform lesion appears in the first few years of life, although in some patients the fundus and visual acuity remain normal. In some patients, multiple vitelliform lesions may be observed in the posterior pole, a variant described as multifocal Best disease81 (Fig. 6–8). In the vitelliform stage, a large yellow, yolk-like lesion appears in the macula at the level of the RPE. These lesions, which appear in infancy or early childhood, are typically bilateral and symmetrical. Visual acuity is usually normal at this stage. In the scrambled-egg stage, there is disintegration of the vitelliform material with irregular subretinal deposits. Visual acuity may be reduced to the 20/30 to 20/40 level. The pseudohypopyon stage occurs when the yellow material gravitates inferiorly in the subretinal space. Areas of RPE atrophy and pigment clumping may be observed. This stage is frequently reached in the teen years.
Lesions Associated with a Flat or Minimally Elevated Retinal Surface
6
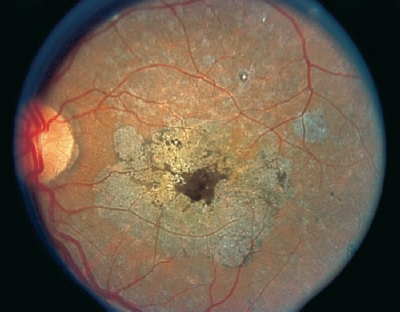
Areas of Fundus Whitening: White or Yellow Spots
Acute Posterior Multifocal Placoid Pigment Epitheliopathy
Who Is Affected?
What Symptoms Are Typical?
What Are Possible Associated Systemic Features?
What Are the Characteristic Ocular Manifestations?
What Does Angiography Show?
What Is the Course of the Disease?
Is There Any Treatment?
Multiple Evanescent White-Dot Syndrome
Who Is Affected?
What Symptoms Are Typical?
What Are Possible Associated Systemic Features?
What Are the Characteristic Ocular Manifestations?
What Does Angiography Show?
Are Other Tests Helpful?
What Is the Course of the Disease?
Is There Any Treatment?
Birdshot Retinochoroidopathy
Who Is Affected?
What Symptoms Are Typical?
What Are the Characteristic Ocular Manifestations?
What Does Angiography Show?
Are Other Tests Helpful?
What Is the Course of the Disease?
Is There Any Treatment?
Stargardt Disease (Fundus Flavimaculatus)
Who Is Affected?
What Symptoms Are Typical?
What Are the Characteristic Ocular Manifestations?
What Does Angiography Show?
Are Other Tests Helpful?
What Is the Course of the Disease?
Is There Any Treatment?
Age-Related Maculopathy
Who Is Affected?
What Symptoms Are Typical?
What Are the Characteristic Ocular Manifestations?
What Does Angiography Show?
What Is the Course of the Disease?
Is There Any Treatment?
Basal Laminar (Cuticular) Drusen
Who Is Affected by Basal Laminar Drusen?
What Symptoms Are Typical?
What Are Associated Systemic Features?
What Are the Characteristic Ocular Manifestations?
What Does Angiography Show?
Are Other Tests Helpful?
What Is the Course of the Disease?
Is There Any Treatment for Basal Laminar Drusen?
Best Disease (Vitelliform Dystrophy)
Who Is Affected?
What Symptoms Are Typical?
What Are the Characteristic Ocular Manifestations?
Ento Key
Fastest Otolaryngology & Ophthalmology Insight Engine