7
Areas of Fundus Whitening: Geographic Whitening
What Is Fundus Whitening, and Why Does It Exist?
Caucasians often have orange–yellow fundi, whereas persons with darker skin frequently have reddish brown fundi. The funduscopic color is a result of the pigment distribution in choroidal melanocytes and the retinal pigment epithelium (RPE). In addition to proper pigment distribution, a fundus of normal color also implies that the three main components of the fundus, the neurosensory retina, RPE, and choroid, are intact and functioning properly. Disruptions in pigmentary or structural components of the fundus may lead to color changes, including retinal whitening. Fundus whitening is often abnormal and may indicate the presence of ischemia, deposition of toxic or metabolic compounds (in the RPE or choroid), infection, or degeneration.
The macula contains xanthophyll (yellow) pigment. The foveal avascular zone (FAZ), a small, slightly concave area devoid of retinal capillaries and occupied by cones, is in the center of the macula. The anatomic fovea and foveola are contained within the center of the FAZ. The central retinal artery, derived from the ophthalmic artery, and its branches are located in the inner retina and supply circulation to all of the inner retinal layers, extending as posteriorly as the inner portion of the inner nuclear layer. The outer retina, from the outer portion of the inner nuclear layer to the RPE, is supported by the choriocapillaris. In some eyes, a cilioretinal artery, which branches from the ciliary circulation, supplies circulation to a portion of the inner retina between the optic nerve and center of the macula.1
The RPE is a single layer of cuboidal cells derived from neuroectoderm. The RPE extends from the margin of the optic disc to the ora serrata and is continuous with the pigment epithelium of the ciliary body. It is phagocytic and continually ingests membranes and disks shed by the outer segments of the photoreceptor cells. This process of shedding, phagocytosis, and photoreceptor renewal follows a daily rhythm. Some pathologic changes occur when the process of phagocytosis and renewal is impaired or affected by genetic defects, senescence, drugs, or dietary insufficiency1 and may be affected by changes in color.
Other pathologies may occur when the RPE is affected or stimulated, leading at times to RPE, hyperplasia, migration, atrophy, and metaplasia. These changes may account for hyperpigmentation or geographic whitening.1
The choroid, a densely pigmented and richly vascularized layer, encases and nourishes the outer retina and RPE. The choroidal circulation is separate from the retinal circulation and is supplied by the short and long posterior ciliary arteries and anterior ciliary arteries. Choroidal arteries branch and decrease in diameter to form a monolayer of capillaries, the choriocapillaris. The four vortex veins located in the midperiphery drain blood from the choroid and anterior uvea. The stroma of the choroid has collagenous and elastic tissue with a variable number of melanocytes. The pigment in these melanocytes and in the RPE contributes to the orange color of the posterior segment. Thus, when there are disturbances in the choroid, such as in types of hereditary choroidal diseases or inflammatory conditions, the fundus changes its normal color and frequently appears to whiten.1
This chapter considers some of the conditions that may present as geographic fundus whitening. In cases of geographic fundus whitening, a thorough medical and ocular history of the patient is important to discern the class of disease: vascular diseases, choroidal dystrophies and degenerations, retinal dystrophies and degenerations, bull’s-eye maculopathy; tumors, infections, uveitis, or idiopathic entities. Once the class of disease is identified, a differential diagnosis associated with that category can be made.
It may be challenging to determine the correct diagnosis for geographic fundus whitening, but the process is facilitated by a carefully inquisitive and systematic approach.
What Are the Major Types of Fundus Whitening?
There are two major types of fundus whitening: (1) white or yellow spots and (2) geographic areas. The differential diagnosis for white or yellow spots is extensive and includes immunologic, infectious, and toxic causes and is addressed in Chapter 6.
What Can Cause Geographic Fundus Whitening?
Idiopathic, vascular, hereditary, toxic, metabolic, immunologic, and degenerative causes can present clinically as geographic fundus whitening. Careful recognition and identification of the disease location, extensive review of the patient’s history and clinical course, and the proper use of diagnostic tools are necessary for proper diagnosis and management.
The appearance of the whitening is helpful in the differential diagnosis. Myelinated nerve fibers, for example, will block the underlying retinal vessels and atrophy, permitting visibility of the choroid, such as choroideremia, gyrate atrophy, and central areolar choroidal dystrophy will permit visibility of the overlying retinal vasculature. Associated signs such as vitreous cells may lead the clinician to the diagnosis of an infectious or inflammatory condition such as acute retinal necrosis (ARN).
Idiopathic Retinal Phenomena That Can Present as Geographic Whitening
Some funduscopic findings are “whitish” in appearance but are not linked to specific causes. Frequent observations include peripheral cobblestone degeneration, nonspecific chorioretinal atrophy, history of previous cryotherapy, and white with and without pressure. Among these, white with and without pressure are the most common incidental findings (Table 7–1).
White With Pressure, White Without Pressure
WHAT ARE THESE CHANGES?
These terms describe geographic areas of whitening in the midperipheral, equatorial, and peripheral retina. During indirect ophthalmoscopy without scleral depression, these areas are described as white without pressure. When a white reflex is detected with scleral depression, it is termed white with pressure.
WHAT IS THE SIGNIFICANCE OF THESE OBSERVATIONS?
The phenomena were described by Schepens in 1952 and initially were considered to predispose to retinal tears.2 Freeman noted an increased risk for giant retinal tears in fellow eyes with white without pressure at the vitreous base.3 Since then, no prognostic or diagnostic significance has been assigned to these reflexes.4 White, with and without pressure, can be detected with coincidental posterior vitreous detachment, thought to represent an equatorial area where the vitreous remains attached to the retina.5 It may also be present in eyes without apparent vitreous detachment. The nature and significance of these phenomena remain controversial.

WHAT MAY BE THE PATHOPHYSIOLOGIC MECHANISM OF THESE FINDINGS?
A light reflex results when the incident light is tangential to more dense bundles of vitreous collagen; this can lead to the appearance of white both with and without pressure.6 Reflection of light from the collagen bundles can cause a white reflex or no reflex, depending on the orientation of the fiber bundles. Therefore, during examination with scleral depression, white or no white may be visible, depending on how scleral indentation distorts the globe and affects the reflex from the collagen fibers.6
HOW ARE THESE FINDINGS TREATED?
Meticulous examination of the retina with scleral depression is indicated. In addition, careful documentation of such findings should be made in the medical records for future reference. Patients should have follow-up examinations every 6 to 12 months or sooner, if new symptoms of vitreous traction are experienced.
Retinal Vascular Diseases That Can Present as Geographic Whitening
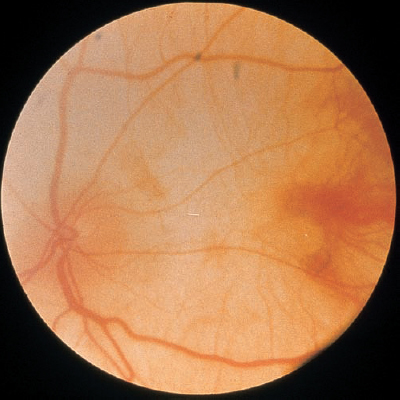
Retinal tissues whiten when there is a lack of blood supply associated with arteriolar occlusion or ischemia. If the retina is damaged and opacified, the white color may persist even after perfusion is reestablished. Over time, the whitening fades as the inner retinal layers atrophy and become thinner. The three most common vascular diseases that lead to retinal whitening are ocular ischemic syndrome and central and branch retinal artery occlusion (Fig. 7–1).
Ocular Ischemic Syndrome
WHAT IS OCULAR ISCHEMIC SYNDROME?
Ocular ischemic syndrome (OIS), described by Kearns and Hollenhorst,7 most often results from severe carotid artery obstructive disease or thrombosis. It has also been called ischemic ocular inflammation8 and ischemic coagulopathy.9,10
WHO IS AT RISK FOR OCULAR ISCHEMIC SYNDROME?
There is no racial predilection associated with OIS, but men are affected more frequently than women. OIS often affects older persons; patients usually are diagnosed in the fifth to the eighth decade of life.11 Either eye can be affected; bilateral ocular involvement occurs in 20% of patients.11 Generally, 90% or greater stenosis of the ipsilateral carotid arterial system is present in eyes with OIS.10 Often, a 90% carotid stenosis is required to reduce the perfusion pressure of the ipsilateral central retinal artery perfusion pressure by about 50%.12,13
WHAT ARE THE OCULAR MANIFESTATIONS OF OCULAR ISCHEMIC SYNDROME?
Both the anterior and posterior segment are involved in OIS. Anterior segment manifestations include rubeosis iridis, neovascular glaucoma, and uveitis (cells and flare). Rubeosis iridis occurs in up to 67% of eyes with OIS at the time of presentation and often is accompanied by flare.10 Only 20% of eyes experience actual anterior chamber cellular inflammatory response, however.10 Only 50% of eyes with OIS and rubeosis iridis have or develop elevated intraocular pressure, even if the anterior chamber angle is closed by fibrovascular tissue.10 Neovascular glaucoma is seen in 35% of eyes with OIS.10
Posterior segment manifestations of OIS can include narrowed retinal arteries (in greater than 90%), dilated retinal veins (in greater than 90%), retinal hemorrhages (80%), neovascularization of optic disc (35%) and retina (8%), cherry-red spot (12%), cotton-wool spots (6%), spontaneous retinal arterial pulsations (4%), vitreous hemorrhages (4%), cholesterol emboli (2%), and ischemic optic neuropathy (2%).10
Most patients (more than 90%) with OIS have a history of visual loss in the affected eye(s). In two thirds of cases, the visual loss is gradual, whereas in 12% of patients, the visual loss is sudden. In cases of abrupt visual loss, a cherry-red lesion is often detected on funduscopic examination.10,11
In as many as 40% of cases, pain in the affected eye or orbital region is present. The pain, termed ocular angina, often is described as a dull ache.10 The visual acuity of OIS patients varies tremendously at presentation, from as good as 20/20 to finger counting or worse.
WHAT CAUSES THE CHERRY-RED SPOT IN OCULAR ISCHEMIC SYNDROME?
Retinal whitening occurs secondary to ischemia, usually caused when the intraocular pressure exceeds the perfusion pressure within the central retinal artery, especially in eyes with neovascular glaucoma,10 or less frequently from embolic obstruction in the central retinal artery. The fovea with residual perfusion from the underlying choroid can remain red and appears as a “cherry-red spot” in comparison with the surrounding white retina.
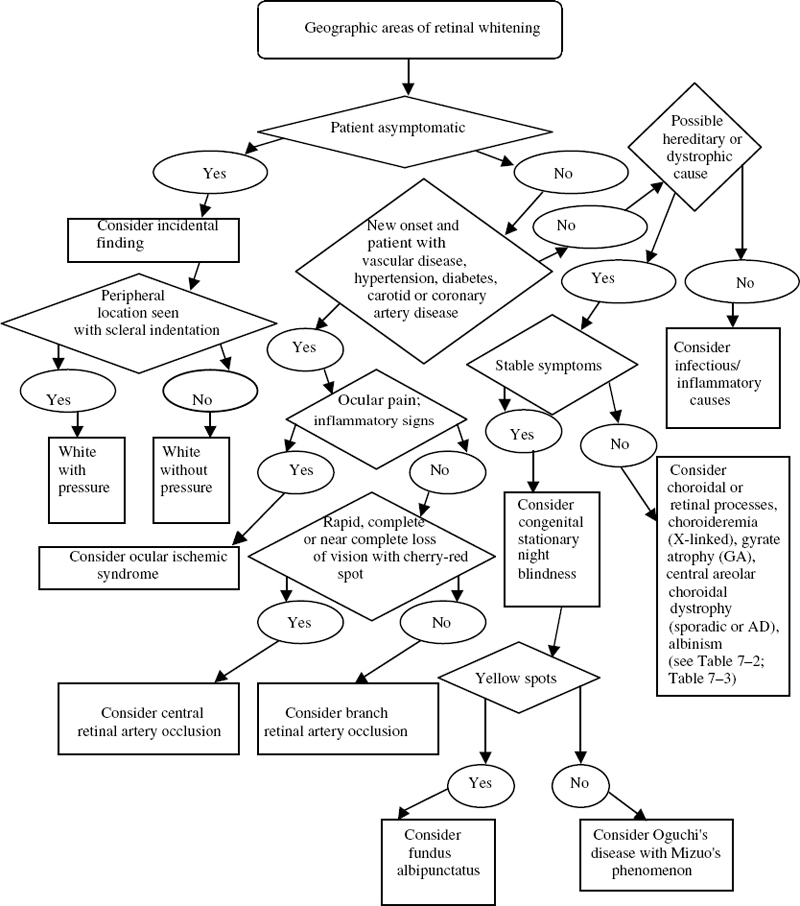
FIGURE 7–1. Clinical pathway: Geographic areas of retinal whitening. AD, autosomal dominant.
HOW IS OCULAR ISCHEMIC SYNDROME TREATED?
The diagnosis of OIS should be established and confirmed with ancillary studies such as fluorescein angiography, which can show a well-demarcated, leading edge of fluorescein dye within a retinal artery,11 or electroretinography (ERG), which may reveal a diminution or absence of the amplitude of the A- and B-waves.10 Carotid artery imaging is a required study. Duplex ultrasonography has less potential for serious complications and thus is preferred over carotid angiography.14 In addition, ophthalmologists should correspond with primary care providers to arrange systemic evaluations for peripheral vascular diseases, cardiovascular diseases, hypertension, and diabetes.
Carotid endarterectomy is recommended and indicated when there is significant carotid disease. When there is 100% carotid artery obstruction, extracranial to intracranial bypass surgery, usually from the superficial temporal artery to the middle cerebral artery, may yield some benefits.11 Medical therapy should be used to lower the intraocular pressure. After ipsilateral carotid endarterectomy, ocular hypertension can occur, which may require, in addition to medical therapy, ciliary body destructive procedures or glaucoma filtering surgery to control. In ocular ischemic eyes with rubeosis iridis and posterior segment neovascularization, panretinal laser photocoagulation may be required.15
Central Retinal Artery Occlusion
WHO IS AT RISK FOR CENTRAL RETINAL ARTERY OCCLUSION?
Both men and women suffer from central retinal artery occlusion (CRAO); however, men are affected more frequently. The incidence of CRAO is estimated to be 1 per 10,000 outpatient visits.16,17 The mean age at the time of presentation is in the sixth decade. Bilateral involvement occurs in 1 to 2% of cases.18
WHAT ARE THE OCULAR MANIFESTATIONS?
Most patients with acute CRAO have a history of abrupt, painless visual loss. Unlike in OIS, the anterior segment is usually normal. If rubeosis iridis is detected at the time the obstruction occurs, the presence of concomitant carotid artery occlusion and a diagnosis of OIS should be considered.17
In acute CRAO, whitening occurs as a result of ischemic necrosis in the affected inner half of the retina.17 The superficial retina becomes opacified and white-yellow, except in the foveolar region, where a cherry-red spot is present (Fig. 7–2). As in OIS, the cherry-red spot can vary in size, depending on the width of the foveola.
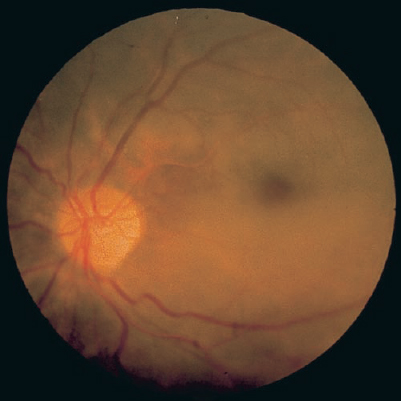
FIGURE 7–2. Central retinal artery occlusion (CRAO) in a 68-year-old woman with coronary artery disease and hypercholesterolemia. The retina has become opacified and has taken a white-yellow appearance, except in the foveolar region, where a cherry-red spot is present.
WHAT CAUSES THE CHERRY-RED SPOT?
In CRAO, most of the retina outside of the macula whitens secondary to ischemia; however, circulation to the foveolar region is partially supplied by the underlying choroid, thereby preventing complete ischemia when the central retinal artery is occluded. The foveolar region retains its reddish hue within the surrounding whitened retina, producing the cherry-red spot.17 Retinal pigmentary changes may be seen if the choroidal circulation is involved. Segmentation of the blood column, or “boxcarring,” may occur in arteries and veins when there is severe obstruction.17
WHAT IS THE VISUAL PROGNOSIS?
Often, CRAO has severe visual morbidity. More than 90% of eyes with CRAO have visual acuity ranging between finger counting to light perception on initial presentation.18 The presence of an embolus in the fundus usually is associated with poor vision. In about 25% of eyes with acute CRAO, there is a patent cilioretinal artery that supplies part or all of the papillomacular bundle.19 If only part of the papillomacular bundle is spared, the visual acuity remains poor and is often no better than 20/100. If the patent cilioretinal artery spares the foveolar region, which occurs in approximately 10% of eyes, visual acuity can be maintained at 20/50 or better.17,19 Initially, patients retain little central vision; however, a large degree of peripheral field often returns.17
HOW IS CENTRAL RETINAL ARTERY OCCLUSION TREATED?
There is no proven treatment for CRAO. The inner retina is highly sensitive due to loss of perfusion; thus, intervention usually is not attempted in any patient with an obstruction more than 72 hours old.
When a patient with acute CRAO is seen within 24 hours after the onset of visual loss, ocular massage can be attempted digitally with a Goldmann contact lens.20 Administration of carbogen, (a mixture of 95% oxygen and 5% carbon dioxide) and anterior chamber paracentesis yield moderate success in some cases.21 Some studies suggest that a high concentration of oxygen, such as with a hyperbaric oxygen chamber, may improve visual function.22 Antifibrinolytic agents have been used,23 although systemic complications, such as cerebrovascular accidents, can occur with this treatment. Systemic anticoagulants are not usually used.24
Branch Retinal Artery Occlusion
WHAT ARE THE CHARACTERISTICS OF BRANCH RETINAL ARTERY OCCLUSION?
Branch retinal artery occlusion (BRAO) occurs in 38% of acute retinal artery occlusion cases,25 compared with CRAO (57%) and cilioretinal artery obstruction (5%). Based on clinical observations, the temporal retinal vessels are often involved in BRAO (in more than 90% of cases).25 Nasal branch retinal arterial obstructions also commonly occur, but they often go unnoticed because they are asymptomatic.17
WHAT ARE THE OCULAR MANIFESTATIONS?
Branch retinal artery occlusion appears as a localized region of superficial retinal whitening, most prominent in the posterior pole, along the distribution of the obstructed or occluded vessel.17 Axoplasmic flow in the nerve-fiber layer is blocked as it reaches the hypoxic retina; this blockage leads to further whitening at the borders of ischemic areas.17
WHAT IS THE VISUAL PROGNOSIS?
The visual prognosis in eyes with BRAO is much better than that of CRAO if the central foveola is not involved and not surrounded by retinal whitening. About 80% have a visual acuity of 20/40 or better, although residual field defects may remain.16 There is no treatment for BRAO.
Hereditary Choroidal Diseases That Can Present as Geographic Whitening
Hereditary choroidal dystrophies can present with focal or generalized regions of choroidal whitening secondary to a degenerative process. Often, the initial diagnosis is made by physical examination; patients may have decreased central vision and a visual field defect. Ophthalmoscopy and fluorescein angiography usually demonstrate bilateral fundus abnormalities. Electroretinogram (ERG) and electrooculogram (EOG) may not be affected until the latter stages of the diseases. Hereditary choroidal dystrophies can be categorized into localized dystrophies (central areolar choroidal dystrophy, Sorsby fundus dystrophy, peripapillary choroidal dystrophy, progressive bifocal chorioretinal atrophy, and Bietti crystalline retinopathy) and generalized choroidal dystrophies (choroideremia and gyrate atrophy) (Table 7–2).26
Gyrate Atrophy
Gyrate atrophy is a type of progressive disorder associated with chorioretinal degeneration and autosomal recessive inheritance. It was first described as an atypical form of retinitis pigmentosa.26 Gyrate atrophy is rare, reported to have an incidence of only one case per 50,000 individuals in Finland.27
| History of previous cryotherapy |
| Bietti crystalline retinopathy |
| Sorsby fundus dystrophy |
| Progressive bifocal chorioretinal atrophy |
| Peripapillary choroidal dystrophy |
| Kearns-Sayre syndrome |
| Age-related macular degeneration |
| Stargardt disease |
| Best disease |
| Pattern dystrophies |
| North Carolina dystrophy |
WHAT ARE THE CLINICAL MANIFESTATIONS OF GYRATE ATROPHY?
Nyctalopia, high myopia, and astigmatism are most often detected during the first decade. Posterior subcapsular cataracts usually develop by the second decade.27,28 The disease continues to progress into the fourth and fifth decades with increasingly constricted visual fields and subsequent loss of central visual acuity.27,28
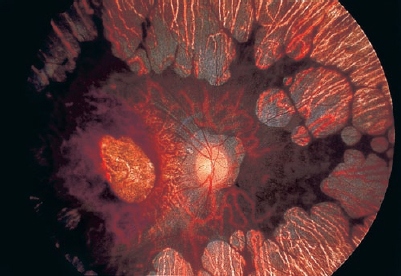
Circular, well-circumscribed areas of chorioretinal atrophy in the midperipheral retina are the hallmarks of gyrate atrophy. These areas enlarge and coalesce in a whitish, scalloped pattern (Fig. 7–3) spreading anteriorly and posteriorly. As the disease progresses to the macula, central vision is threatened.26 Although gyrate atrophy is in the differential diagnosis of retinal whitening, its unique pattern of lesions is distinctive; thus, gyrate atrophy often is diagnosed clinically.
WHAT IS THE GENETIC DEFECT IN GYRATE ATROPHY?
Patients with gyrate atrophy have been found to have plasma ornithine levels 10 to 20 times higher than those of controls, secondary to a deficiency in ornithine aminotransferase (OAT),29 with two thirds of patients having no detectable OAT activity.26,30 Other biochemical abnormalities include hyperornithinemia and reductions in plasma lysine, creatine, glutamine, and glutamate.26,30
The gene for OAT has been linked to chromosome 10q26 and has been cloned.31 There have been more than 60 distinct mutations described in gyrate atrophy.26,32
HOW IS GYRATE ATROPHY TREATED?
Therapy has been geared to reducing plasma ornithine levels. In a small number of patients with the pyridoxine-responsive form of gyrate atrophy, supplemental pyridoxine reduces ornithine levels, although the effects on visual prognosis are not known.33 In some patients, diets low in arginine, the precursor of ornithine, have been shown to delay the progression of visual deterioration.34
Choroideremia
Choroideremia26 is an X-linked recessive disorder characterized by the progressive degeneration of the RPE, retina, and choroid.35 In the first and second decades of life, patients present with problems with dark adaptation. By the sixth or seventh decade of life, patients may have varying degrees of central visual acuity loss and severe constriction of visual fields.26
WHAT IS THE GENETIC DEFECT IN CHOROIDEREMIA?
The gene for choroideremia has been linked to chromosome Xq13-q22 and cloned.36 The protein product of this gene in humans has been found to be identical to rat Rab escort protein (REP1), which is required for the function of Rab geranylgeranyl transferase in intracellular vesicular transport.37 Although multiple mutations have been discovered in this gene, including deletions and translocations, they all result in protein truncation.38
WHAT ARE THE CLINICAL MANIFESTATIONS OF CHOROIDEREMIA?
Areas of pigment stippling and whitish focal atrophy of the RPE are initial fundus abnormalities, seen as early as the first decade in affected males and asymptomatic female carriers. The early features are similar to those seen in retinitis pigmentosa. With disease progression, choroidal atrophy increases, causing areas of whitening from greater visibility of the sclera. There is also photoreceptor and RPE loss, exposure of the choroidal vessels, and sparing of small areas of choroid in the macula and periphery26 (Fig. 7–4). Visual function is severely impaired. There is no treatment to prevent or reverse choroideremia at present.
Central Areolar Choroidal Dystrophy
Central areolar choroidal dystrophy is a slowly progressive bilateral disease beginning in the second to fourth decade of life.39 Most cases are sporadic, but some have autosomal-dominant inheritance. One gene has been localized to chromosome 17p25.40