Chapter 32 Anterior segment
developmental anomalies
The anterior segment of the eye is an intricate arrangement of interacting tissues essential for vision. The cornea, iris, and the anterior epithelium of the lens form the boundaries of the anterior chamber. Schwalbe’s line, the trabecular meshwork, and the scleral spur lie in the anterior chamber angle at the junction of the peripheral cornea and the root of the iris. Aqueous produced by the ciliary body flows into the anterior chamber through the pupil and leaves the eye through the trabecular meshwork into Schlemm’s canal and the venous circulation.1
Embryology of the anterior segment
In addition to the large contribution from neural crest-derived mesenchymal cells (Table 32.1) to tissues of the anterior segment, the neurectoderm of the optic cup and the surface ectoderm also give rise to anterior segment components. The peripheral edge of the optic cup forms the posterior iris epithelium and the ciliary body epithelium. The surface ectoderm gives rise to the corneal epithelium after the separation of the lens vesicle. Please see Fig. 32.1 for a brief reminder of embryologic development of the eye.
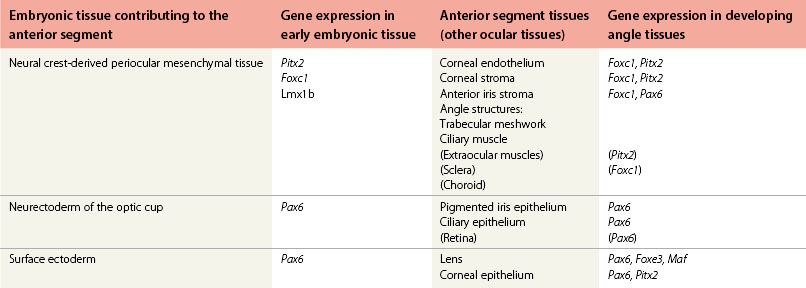
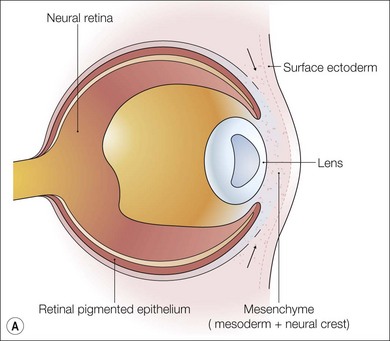
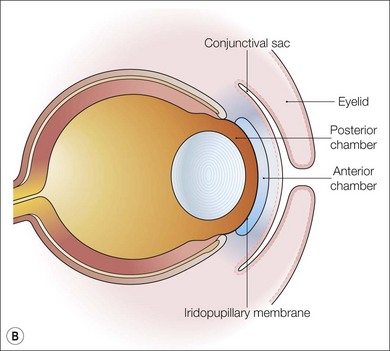
Fig. 32.1 Early development of the anterior segment. (A) By five weeks of development in the human embryo, the lens vesicle has separated from the surface ectoderm and neural crest cells are migrating around the optic cup and between the surface ectoderm and the developing lens. (B) During the seventh week, the mesenchymal layer gives rise to the corneal stroma, bounded by an endothelium, and the anterior iris stroma. This process of differentiation occurs simultaneously with a separation of the two layers to form the anterior chamber between the developing cornea and the iris. (C) A sheet of mesenchyme bridging the future pupil remains until the seventh month of gestation. The edges of the optic cup form the posterior iris epithelium and the ciliary body epithelium.2,3
Control of development: responsible genes
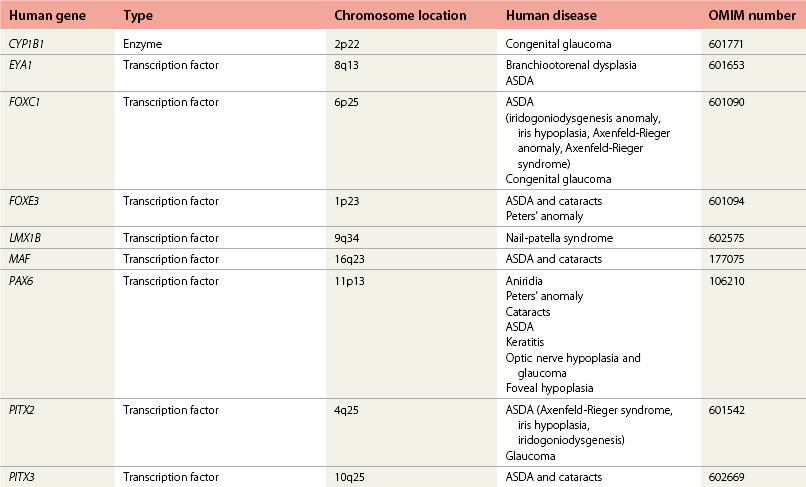
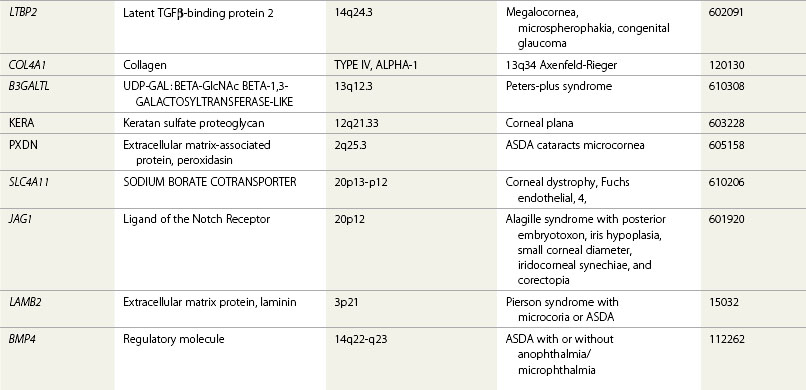
Gene mutations causing anterior segment developmental anomalies
Several disease causing genes have been identified (Table 32.2) and animal models have illuminated the essential role of these genes in the normal molecular and cellular processes underlying the development of the anterior segment.
Transcription factors and anterior segment development
The majority of genes proven to play critical roles in anterior segment developmental anomalies (ASDAs) all encode transcription factors (Table 32.2), which act by regulating the transcription of other genes, which are their downstream target genes. They coordinate programs of growth and differentiation by either enhancing or repressing the expression of their target genes. Each transcription factor has a different type of DNA-binding domain for the purpose of interacting with the regulatory DNA sequence of its target genes. These DNA-binding domains are highly conserved throughout the animal kingdom, and this is partly why animal models have proved to be so useful for understanding the function of human genes. PAX6 and PITX2 encode proteins containing a paired-type and bicoid-type homeodomain, respectively, whereas FOXC1 encodes a fork-head domain containing protein. These genes are closely related to genes called paired, bicoid, and forkhead, first shown to be essential for development and patterning the body of Drosophila melanogaster, an invaluable model system for the discovery of many genes important for human development and disease.
Gene expression in the developing anterior segment: sites of gene action
The sites of gene expression during development of the anterior segment pinpoint their site of action (Fig. 32.1 and Table 32.1). Knowledge of sites of gene expression derives mainly from study of the mouse as a model for mammalian development combined with limited expression data from human studies and other animal model or experimental systems. This information helps understand the origin of the abnormalities observed in patients with gene mutations.
The patterns of expression of genes whose mutation causes ASDAs can be considered as three types:
1. Those expressed within migrating periocular neural crest cells (Foxc1, Pitx2, Lmx1b)
2. Those expressed only within the developing lens (Foxe3, Maf)
3. Those with a more panocular expression including the lens, of which Pax6 is the only example.
Foxc1 and Pitx2 are essential for corneal development
Developmental arrest and abnormal retention and contraction of the embryonic endothelial layer on portions of the iris and anterior chamber angle have been proposed as the cause of the iris changes, tissue strands, and the abnormalities of Schwalbe’s line found in ASDAs.9
Heterozygous mutation of the Foxc1 gene in mice causes ocular abnormalities with marked variable expressivity, which are very similar to patient conditions and the widely variable ocular defects sometimes seen within families sharing the same mutation.10,11 Ocular defects in the heterozygotes are progressive with a gradual worsening of the corectopia and with the peripheral iridocorneal adhesions becoming more evident over time. Some juvenile mice showed mild corneal opacity progressing to a high incidence of corneal opacification, with neovascularization and cataracts, in older animals.
Mice that are homozygous for mutation of Foxc1 (called the congenital hydrocephalus, ch, or Mf1 gene-targeted mouse) show a more severe dysgenesis of the anterior segment.4,12 Histologic analysis shows that the cornea fails to separate from the lens, resulting in the complete absence of an anterior chamber. The outer corneal epithelium is thicker than normal and the stroma is disorganized. There is no differentiation of the inner corneal endothelial layer, and there is a failure to form tight occluding junctions between these posterior endothelial cells necessary for normal physiologic barrier function. Descemet’s membrane, the basal lamina secreted by the corneal endothelium, is thus absent. Hypoplasia of the iris stromal mesenchyme and the pigmented layer accompanies the corneal abnormalities, and the eye is typically microphthalmic. The phenotype of these mice suggests that Foxc1 is essential for conversion of mesenchymal neural crest cells to an endothelium phenotype.
The phenotype of the mice homozygous for Foxc1 mutation is strikingly similar to that of mice carrying homozygous mutation of another gene implicated in human ASDAs, the Pitx2 gene. Mice homozygous for mutation of Pitx2 have displaced, irregular pupils, and this condition is also present in some heterozygotes. In homozygotes, the anterior chamber and the corneal endothelium are absent, and the corneal epithelium is thickened (hypercellular) with undifferentiated mesenchymal cells lying between this epithelium and the optic cup.13 Pitx2 appears essential for differentiation of both the mesenchymal and epithelial components of the cornea, tissues derived from the cranial neural crest-derived periocular mesenchyme and the surface ectoderm, respectively. Pitx2 expression, but not Foxc1 expression, has been reported in the corneal ectoderm (derived from the surface ectoderm).5 Lack of Pitx2 in mice also causes failure of extraocular muscle development, reduced eye size (microphthalmia), and delay in optic fissure closure (optic nerve coloboma). These conditions have not been observed in patients with PITX2 mutation.
Mice that lack Pitx2 also have abnormalities in multiple organs that are essential sites of Pitx2 gene activity. These include roles for Pitx2 in left–right asymmetry involved in cardiac positioning and lung asymmetry and pituitary, craniofacial, and tooth development. Only eye and tooth abnormalities are apparent in heterozygote animals,14 and these are consistent with the dental abnormalities found in patients with PITX2 mutation. Knowledge of other organs critically affected by lack of Pitx2 is useful for understanding other systemic features often identified in patients with anterior segment dysgenesis. Recently it has been shown that complex severe ASDA phenotypes may be due to digenic inheritance of PITX2 and FOXC1 in humans.15
Pax6 and other genes expressed in the developing lens cause ASDAs
It is now well established that mutation or deletion of the PAX6 gene and/or chromosomal rearrangements involving the PAX6 gene on 11p13 underlies most cases of aniridia.6,16 PAX6 is also more widely implicated in anterior segment malformations. Mice with heterozygous mutations of the Pax6 gene help in understanding the role of Pax6 in ASDA as their phenotype resembles the patient conditions associated with PAX6 mutation. Heterozygous Pax6 (small eye (Sey)) mice have a reduced eye size (microphthalmia) and a wide spectrum of anterior eye defects including iris hypoplasia, iridocorneal adhesions and corneal opacification, incomplete separation of the lens from the cornea (keratolenticular adhesion), vascularized cornea, and cataracts.17,18
Peters’ anomaly (see later), characterized by keratolenticular and/or iridocorneal adhesion, is a genetically heterogeneous condition. A proportion of cases of Peters’ anomaly have PAX6 mutations, and the phenotype of the Sey heterozygous mice resembles that of Peters’ anomaly.19 FOXE3 mutation has been associated with Peters’ anomaly, and mice heterozygous for Foxe3 mutation also have central corneal opacity and keratolenticular adhesion similar to that in Peters’ anomaly.20
The lens plays an essential role in the induction of anterior segment differentiation.21,22 Analysis of heterozygous Pax6 eyes indicates that haploinsufficiency of Pax6 causes primary defects in the lens and that these underlie secondary complex defects of the anterior segment iris and cornea. Pax6 is highly expressed in anterior lens epithelium and may act indirectly on neural crest-derived mesenchymal cells of the developing anterior segment by regulating the production of lens-derived signaling molecules. Two other genes are implicated in causing ASDA by affecting the inductive properties of the lens. MAF and FOXE3 mutation both cause ASDA with cataracts. In the mouse the Maf and the Foxe3 genes are both primarily expressed in the developing lens and not within the neural crest-derived mesenchymal cells of the developing anterior segment. Recent work has shown that homozygous mutations in FOXE3 result in primary congenital aphakia.23
Insights into the etiology of developmental glaucoma from mouse models of ASDA
In Foxc1 homozygous mice, histologic analysis of the iridocorneal angle identified abnormalities, including small or absent Schlemm’s canal, hypoplastic or absent trabecular meshwork, and hypoplastic ciliary body with short and thin ciliary processes.10 The development of the chamber angle has also been studied in Pax6 heterozygotes. Mesenchymal cells at the angle that normally express Pax6 and differentiate into trabecular meshwork cells next to Schlemm’s canal remain undifferentiated, demonstrating that Pax6 is directly required for differentiation of the angle.18
Clinical conditions due to anterior segment developmental anomalies
ASDAs may be considered in terms of their embryologic origin. Therefore they may be:
Anterior segment developmental anomalies of neural crest cell origin
Posterior embryotoxon
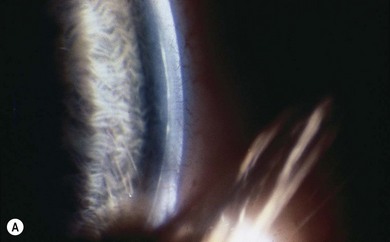
Ocular associations may include iris adhesions with or without iris changes such as hypoplasia, pseudopolycoria, and/or corectopia, in which case it forms part of the spectrum of the Axenfeld-Rieger anomaly (Fig. 32.2).
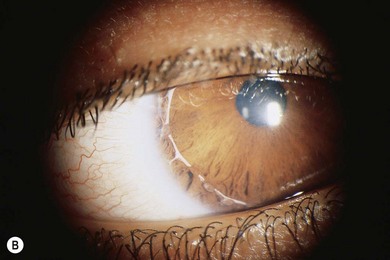
The main systemic association is in a jaundiced neonate, in which case its presence may be suggestive of the autosomal dominant condition Alagille syndrome (arteriohepatic dysplasia)24–26 which is characterized by intrahepatic cholestasis, peripheral pulmonary artery stenosis, peculiar facies, and butterfly vertebral arch defects. Posterior embryotoxon is seen in 90% of all cases and 77% of cases also have iris strands.24,25
Axenfeld-Rieger syndrome (ARS) has ocular and non-ocular features. The ocular features consist of posterior embryotoxon with iris strands attached, some of which may be very broad and thick and others thread-like. Historically if these were the only findings the term Axenfeld anomaly was used. If in addition iris defects are present then historically this was termed Rieger anomaly. Axenfeld-Rieger anomaly encompasses both now. Iris findings range from stromal hypoplasia, pseudopolycoria, corectopia (pupil displaced toward a thick peripheral iris strand) (Fig. 32.3), and ectropion uveae. The anterior chamber angle is usually open though there may be a high insertion of the iris into the posterior portion of the trabecular meshwork.9,27,28,33 Occasionally the pupil corectopia is severe enough to warrant surgical pupilloplasty. The pupil may still progress over years to become even more eccentric in placement despite surgery.
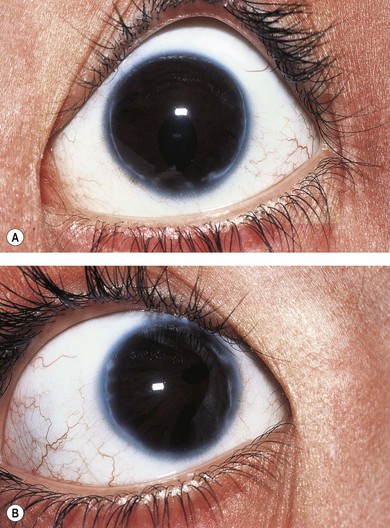
Angle and iris changes are usually stable. Glaucoma develops in 50−60% of patients with ARS, usually manifesting itself in childhood or young adulthood. Incomplete development of the trabecular meshwork and Schlemm’s canal is thought to occur again due to development arrest occurring during the third trimester, causing obstruction to aqueous outflow and hence glaucoma.9,28
Familial glaucoma iridogoniodysplasia has been described in one pedigree and entails marked iris hypoplasia, iridocorneal angle anomalies, and, frequently, glaucoma.29
IGDA has been described as iridocorneal angle anomalies, iris stromal hypoplasia, and glaucoma in 50% of cases.30
IGDS is a rare condition in which iris hypoplasia and iridocorneal angle anomalies are associated with non-ocular features such as jaw and dental abnormalities.31
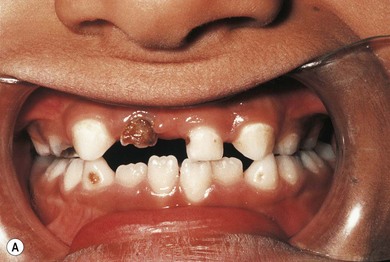
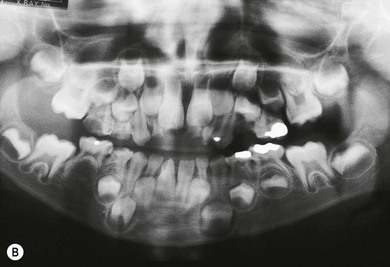
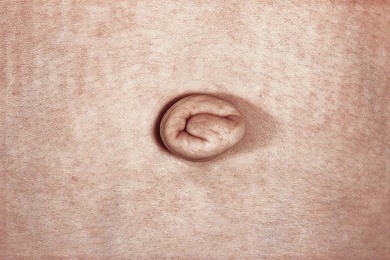
The characteristic non-ocular features of ARS are maxillary hypoplasia, mild prognathism, hypodontia (decreased but evenly spaced teeth), anodontia/oligodontia (focal absence of teeth), microdontia (reduction in crown size), cone-shaped teeth (Fig. 32.4), and excess periumbilical skin (Fig. 32.5) with or without hernia. Hypertelorism, telecanthus, and a broad flat nose have also been described.32 Other systemic features that have been reported include growth hormone deficiency and short stature, heart defects, middle ear deafness, mental deficiency, cerebellar anomalies (FOXC1 mutations), oculocutaneous albinism, hypospadias, abnormal ears, and, in one pedigree, myotonic dystrophy and Peters’ anomaly.9,34
Trabeculectomy with antimetabolite augmentation appears to be the procedure of choice for most patients with glaucoma secondary to ARS, especially in older children (Fig. 32.6). The use of cycloablation should be considered in cases where, because of cooperation of the child, a drainage procedure would be unsuitable. Draining procedures other than trabeculectomy include the use of drainage tubes with or without antimetabolite augmentation.35–38
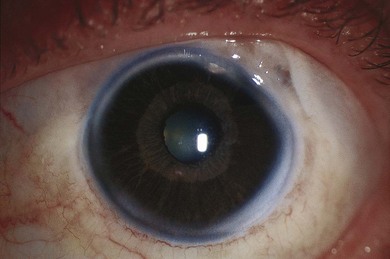
Congenital iris ectropion
This is a rare usually unilateral condition in which there is a congenital, non-progressive, non-tractional hyperplasia of the posterior pigment iris epithelium onto the anterior surface of the iris (ectropion). The child is often thought to have mydriasis and anisocoria because of the dark nature of the posterior pigment epithelium. The ectropion may be circumferential (Fig. 32.7) or sectorial. Other ocular features include iris stromal hypoplasia, a high iris insertion into the trabeculum with goniodysgenesis, and secondary glaucoma.
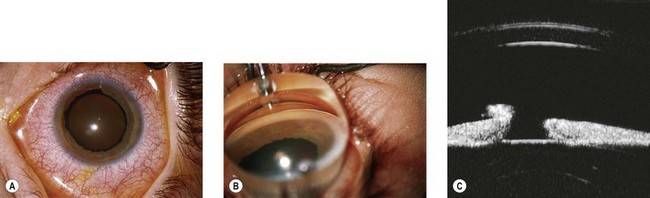
The clinical features are thought to result from an arrest in development with abnormal retention of primordial endothelium, which explains the central iris and angle changes. Although the affected pupil reacts to light and accommodation it may not do so at the same speed as the unaffected eye.7,39,40 Glaucoma occurs in the majority of these patients usually between early childhood and puberty.
Systemic associations that should be excluded include neurofibromatosis I and Prader-Willi syndrome.
Management of the glaucoma can be difficult with medical therapy often being unsuccessful and augmented trabeculectomy is often the surgical operation of choice.7,39,40
Primary congenital glaucoma
ICE syndromes
The iridocorneal endothelial (ICE) syndromes, thought to be due to a primary neural crest cell abnormality, include progressive essential iris atrophy, Chandler syndrome, and the iris-nevus syndrome (also known as Cogan-Reese syndrome).41–43
These rare syndromes are even rarer in children. They are unilateral with females affected more than males and are almost exclusively found in white people. However, specular microscopy almost always reveals mild corneal and iris abnormalities in the “unaffected” eye.44
Specular microscopy studies45 of early cases of ICE syndrome suggest that a subpopulation of corneal endothelial cells are congenitally abnormal and subsequently migrate/proliferate at a slow rate, resulting in a delayed onset of symptoms or diagnosis. These cells may migrate over the angle and onto the iris to cause different signs such as glaucoma and peripheral anterior synechiae. Descriptions of the trabecular meshwork in a 16-year-old with ICE syndrome show collapsed trabecular beams and decreased intertrabecular spaces.46
Progressive iris atrophy shows corectopia, iris ectropion, and pseudopolycoria. Peripheral anterior synechiae often form and gradually become very broad based. Iris holes and thinning occur due to contraction of the membrane formed by the migrating cells. The cornea can appear normal in this condition or have an appearance at the endothelial level similar to that of Fuchs’ dystrophy. Glaucoma is not uncommon in this condition and may develop before extensive iris changes. Chandler syndrome is usually associated with corneal edema due to corneal endothelial changes, but iris changes, if present, are much milder than those seen in progressive essential iris atrophy, with glaucoma if present also being more easily controlled medically. Iris-nevus syndrome consists of iris changes either of a nodular type or a flattish pigmented type. These lesions may be associated with varying degrees of iris atrophy and/or corneal endothelial changes.41–43
Management is purely of any glaucoma that may occur in the first instance. Children are unlikely to require corneal grafting but may do so in their adult lives.47 Glaucoma management should be attempted initially with aqueous production-suppressing topical treatment but filtering surgery may be needed. Usually antimetabolite augmented trabeculectomy is favored, whereas some authors also advocate the use of drainage tubes.
Anterior segment developmental anomalies of ectodermal origin
The two main conditions in this category are limbal and corneal dermoids, which may be associated with Goldenhar syndrome with up to 30% of patients being affected48–50 (see Chapter 29 and Figs 29.2 to 29.4, Figs 32.8 and 32.9).
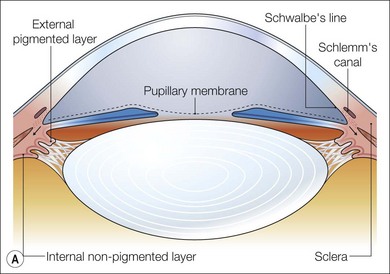
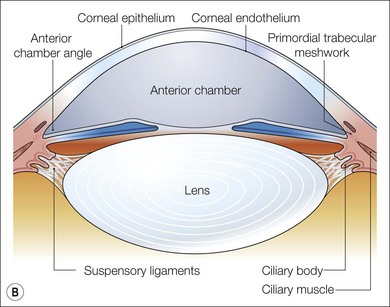
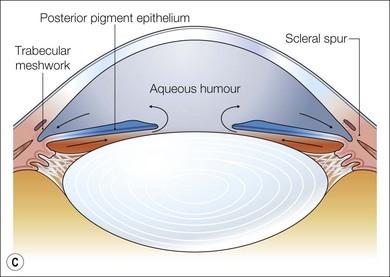
Fig. 32.8 Maturation of the angle of the anterior segment. (A) By 5 months the iris insertion is anterior to the trabecular meshwork primordia. (B) Realignment of the iris insertion gradually uncovers the developing trabecular meshwork. (C) At birth the iris insertion has reached the level of the scleral spur uncovering the angle.7–9
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


