Idiopathic
 Collagen vascular diseases
Collagen vascular diseases
 Rosacea
Rosacea
 Gout
Gout
 Herpes zoster, herpes simplex, syphilis
Herpes zoster, herpes simplex, syphilis
Symptoms
 Acute onset of redness, mild irritation, tearing, rarely, pain
Acute onset of redness, mild irritation, tearing, rarely, pain
Signs
 Simple episcleritis
Simple episcleritis
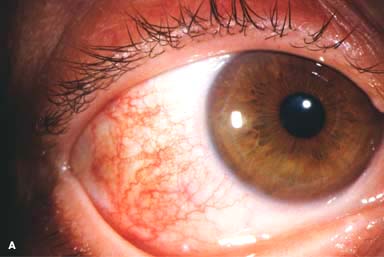
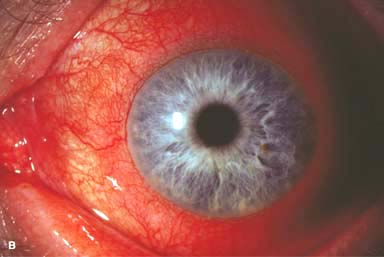
 Sectorial or diffuse hyperemia, primarily involving the middle episcleral plexus, with some secondary involvement of the overlying conjunctival vessels (Fig. 9-1)
Sectorial or diffuse hyperemia, primarily involving the middle episcleral plexus, with some secondary involvement of the overlying conjunctival vessels (Fig. 9-1)
 The inflamed episcleral vessels have a straight, radial configuration.
The inflamed episcleral vessels have a straight, radial configuration.
 Topical phenylephrine 2.5% will cause blanching and enhance visualization of the normal deep vascular plexus over the sclera.
Topical phenylephrine 2.5% will cause blanching and enhance visualization of the normal deep vascular plexus over the sclera.
 Nodular episcleritis
Nodular episcleritis
 Somewhat tender, usually solitary, localized injected nodule that can be moved slightly over the sclera
Somewhat tender, usually solitary, localized injected nodule that can be moved slightly over the sclera
 A localized area of corneal thinning secondary to desiccation (delle) may develop adjacent to an episcleral nodule.
A localized area of corneal thinning secondary to desiccation (delle) may develop adjacent to an episcleral nodule.
Differential Diagnosis
 Conjunctivitis: usually associated with papillary or follicular response in the tarsal conjunctiva. May have a mucoid or purulent discharge
Conjunctivitis: usually associated with papillary or follicular response in the tarsal conjunctiva. May have a mucoid or purulent discharge
 Scleritis: Pain is deeper and more severe, sclera has a purple or bluish hue under natural light, injected scleral vessels have criss-crossing configuration, vessels are immobile over the globe, patients tend to be older.
Scleritis: Pain is deeper and more severe, sclera has a purple or bluish hue under natural light, injected scleral vessels have criss-crossing configuration, vessels are immobile over the globe, patients tend to be older.
 Inflamed pinguecula: located in the completely mobile conjunctiva
Inflamed pinguecula: located in the completely mobile conjunctiva
Diagnostic Evaluation
 Examination is made with the slit lamp and under natural or room light.
Examination is made with the slit lamp and under natural or room light.
 Attempt to move the episcleral vessels over the sclera using a cotton-tipped stick under topical anesthesia.
Attempt to move the episcleral vessels over the sclera using a cotton-tipped stick under topical anesthesia.
 Apply phenylephrine 2.5% and observe for blanching of episcleral vessels after 15 minutes.
Apply phenylephrine 2.5% and observe for blanching of episcleral vessels after 15 minutes.
 Systemic work-up if history is suggestive of collagen vascular disease or gout.
Systemic work-up if history is suggestive of collagen vascular disease or gout.
Treatment
 Artificial tears and cool compresses q.i.d.
Artificial tears and cool compresses q.i.d.
 Consider a short course of a topical NSAID (e.g., diclofenac q.i.d., ketorolac q.i.d., bromfenac q.d. or b.i.d., or nepafenac t.i.d.).
Consider a short course of a topical NSAID (e.g., diclofenac q.i.d., ketorolac q.i.d., bromfenac q.d. or b.i.d., or nepafenac t.i.d.).
 Oral NSAID (e.g., ibuprofen 400 to 600 mg t.i.d. to q.i.d., indomethacin 12.5 to 25 mg q.d. to t.i.d. or flurbiprofen 100 mg b.i.d. to t.i.d.) for moderate to severe cases.
Oral NSAID (e.g., ibuprofen 400 to 600 mg t.i.d. to q.i.d., indomethacin 12.5 to 25 mg q.d. to t.i.d. or flurbiprofen 100 mg b.i.d. to t.i.d.) for moderate to severe cases.
 Consider a short course of topical corticosteroids (e.g., fluorometholone 0.1% to 0.25%, loteprednol 0.2% to 0.5%) q.i.d. in recalcitrant cases. Rarely, oral corticosteroids are required.
Consider a short course of topical corticosteroids (e.g., fluorometholone 0.1% to 0.25%, loteprednol 0.2% to 0.5%) q.i.d. in recalcitrant cases. Rarely, oral corticosteroids are required.
Prognosis
 Good to very good. Episcleritis is often a recurrent condition.
Good to very good. Episcleritis is often a recurrent condition.
Figure 9-1. Episcleritis. A. Localized injection of the episcleral vessels is evident in this patient with sectorial episcleritis. Episcleritis. B. Diffuse injection of the episcleral vessels is present in this eye. Episcleritis can be difficult to differentiate from scleritis in some patients. Episcleral vessels tend to blanch with topical phenylephrine 2.5%, whereas scleral vessels do not.
ANTERIOR SCLERITIS
Scleritis is a severe, potentially sight-threatening ocular disorder that has a totally different prognosis than episcleritis. It may be mild and benign or severe and destructive. Females are affected more often than males, and the condition is frequently bilateral. Most scleritis affects the anterior sclera. Anterior scleritis can be divided into the clinical forms described below.
Classification
 Nonnecrotizing scleritis
Nonnecrotizing scleritis
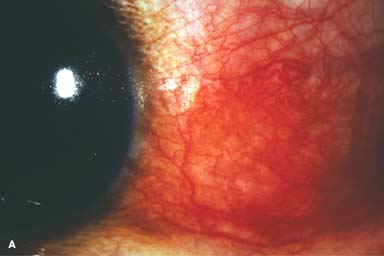
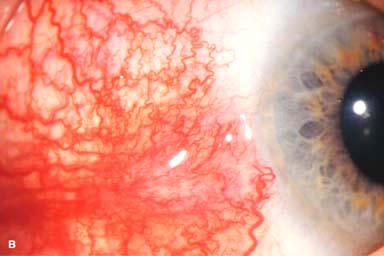
 Diffuse: diffuse hyperemia and distortion of the pattern of the deep vascular plexus, associated with variable episcleral and conjunctival injection (Fig. 9-2A). This is the most benign form and is associated with the least severe systemic conditions.
Diffuse: diffuse hyperemia and distortion of the pattern of the deep vascular plexus, associated with variable episcleral and conjunctival injection (Fig. 9-2A). This is the most benign form and is associated with the least severe systemic conditions.
 Nodular: Tender, usually solitary, deep, localized injected nodule that cannot be moved over the sclera (Fig. 9-2B)
Nodular: Tender, usually solitary, deep, localized injected nodule that cannot be moved over the sclera (Fig. 9-2B)
 Necrotizing scleritis
Necrotizing scleritis
 With inflammation
With inflammation
![]() This is the most destructive form of scleritis. It is associated with ocular or systemic complications in 60% of patients, and 40% may have loss of vision. One-third of patients may die within a few years as a result of severe autoimmune disease if they are inadequately treated.
This is the most destructive form of scleritis. It is associated with ocular or systemic complications in 60% of patients, and 40% may have loss of vision. One-third of patients may die within a few years as a result of severe autoimmune disease if they are inadequately treated.
![]() Gradual appearance of painful, localized, avascular patch overlying an area of scleral necrosis (Fig. 9-2C)
Gradual appearance of painful, localized, avascular patch overlying an area of scleral necrosis (Fig. 9-2C)
![]() Inflammation may be localized to the surrounding sclera or may become diffuse.
Inflammation may be localized to the surrounding sclera or may become diffuse.
![]() The underlying uvea becomes progressively visible through the thinned and necrotic sclera.
The underlying uvea becomes progressively visible through the thinned and necrotic sclera.
 Without inflammation (scleromalacia perforans)
Without inflammation (scleromalacia perforans)
![]() Typically seen in patients with long-standing rheumatoid arthritis
Typically seen in patients with long-standing rheumatoid arthritis
![]() Asymptomatic development of enlarging gray-blue patches of scleral thinning
Asymptomatic development of enlarging gray-blue patches of scleral thinning
![]() Exposure of underlying uvea through areas of thin, devitalized sclera with large bridging vessels
Exposure of underlying uvea through areas of thin, devitalized sclera with large bridging vessels
![]() Anterior scleral staphylomas can occur.
Anterior scleral staphylomas can occur.
Etiology
 Half of patients with scleritis have an associated systemic disease.
Half of patients with scleritis have an associated systemic disease.
 Idiopathic
Idiopathic
 Iatrogenic
Iatrogenic
 Surgery (e.g., cataract surgery, trabeculectomy, scleral buckle) (Fig. 9-2D)
Surgery (e.g., cataract surgery, trabeculectomy, scleral buckle) (Fig. 9-2D)
 Topical medications (e.g., NSAIDs, corticosteroids) (Fig. 9-2E)
Topical medications (e.g., NSAIDs, corticosteroids) (Fig. 9-2E)
 Collagen vascular diseases
Collagen vascular diseases
 Rheumatoid arthritis (most common)
Rheumatoid arthritis (most common)
 Wegener’s granulomatosis (moderately common)
Wegener’s granulomatosis (moderately common)
 Polyarteritis nodosa
Polyarteritis nodosa
 Relapsing polychondritis
Relapsing polychondritis
 Others (e.g., systemic lupus erythematosus, juvenile rheumatoid arthritis, juvenile chronic arthritis, polymyositis)
Others (e.g., systemic lupus erythematosus, juvenile rheumatoid arthritis, juvenile chronic arthritis, polymyositis)
 Granulomatous diseases
Granulomatous diseases
 Sarcoidosis
Sarcoidosis
 Tuberculosis
Tuberculosis
 Syphilis
Syphilis
 Lyme disease
Lyme disease
 Skin diseases
Skin diseases
 Herpes zoster ophthalmicus (moderately common)
Herpes zoster ophthalmicus (moderately common)
 Acne rosacea
Acne rosacea
 Gout
Gout
Complications
 Stromal keratitis, peripheral corneal melt and sclerosing keratitis, associated with worse prognosis
Stromal keratitis, peripheral corneal melt and sclerosing keratitis, associated with worse prognosis
 Uveitis
Uveitis
 Cataract
Cataract
 Glaucoma
Glaucoma
 Posterior uveitis, posterior scleritis, exudative retinal detachment
Posterior uveitis, posterior scleritis, exudative retinal detachment
Symptoms
 Severe, boring pain that may radiate to the orbit or head and awaken patients from sleep; redness; tearing; photophobia; decreased vision
Severe, boring pain that may radiate to the orbit or head and awaken patients from sleep; redness; tearing; photophobia; decreased vision
 Onset is generally gradual but may be acute.
Onset is generally gradual but may be acute.
 History of associated systemic diseases in many cases
History of associated systemic diseases in many cases
Signs
 Inflamed conjunctiva, episclera, and sclera. Scleral vessels have criss-crossing patterns, do not blanch with 2.5% phenylephrine drops, and are immobile over the globe.
Inflamed conjunctiva, episclera, and sclera. Scleral vessels have criss-crossing patterns, do not blanch with 2.5% phenylephrine drops, and are immobile over the globe.
 Sclera has a purple-bluish hue when examined grossly under natural light.
Sclera has a purple-bluish hue when examined grossly under natural light.
 Scleral inflammation can be sectorial or diffuse. Scleral edema, nodule, or thinning and necrosis may be seen. Sometimes, the sclera may become more translucent without significant thinning.
Scleral inflammation can be sectorial or diffuse. Scleral edema, nodule, or thinning and necrosis may be seen. Sometimes, the sclera may become more translucent without significant thinning.
 Anterior chamber reaction
Anterior chamber reaction
 Other signs of ocular complications as mentioned above may be observed.
Other signs of ocular complications as mentioned above may be observed.
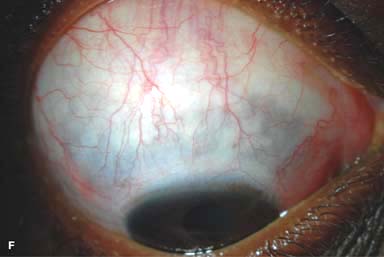
 Episodes of resolved scleritis can result in blue-gray areas of thinned sclera (Fig. 9-2F).
Episodes of resolved scleritis can result in blue-gray areas of thinned sclera (Fig. 9-2F).
Differential Diagnosis
 Episcleritis
Episcleritis
 Inflamed pinguecula
Inflamed pinguecula
 Infectious scleritis
Infectious scleritis
Diagnostic Evaluation
 Examine with the slit lamp and under natural or room light.
Examine with the slit lamp and under natural or room light.
 Attempt to move the injected vessels over the sclera using a cotton-tipped stick under topical anesthesia.
Attempt to move the injected vessels over the sclera using a cotton-tipped stick under topical anesthesia.
 Apply phenylephrine 2.5% and observe for absence of blanching of the scleral vessels after 15 minutes.
Apply phenylephrine 2.5% and observe for absence of blanching of the scleral vessels after 15 minutes.
 Consider cultures if there is an overlying conjunctival epithelial defect.
Consider cultures if there is an overlying conjunctival epithelial defect.
 Investigate for associated systemic diseases.
Investigate for associated systemic diseases.
Treatment
 Nonnecrotizing diffuse and nodular scleritis
Nonnecrotizing diffuse and nodular scleritis
 Oral NSAIDs (e.g., indomethacin
Oral NSAIDs (e.g., indomethacin
25 mg t.i.d. or flurbiprofen 100 mg t.i.d.) are often effective.
 For more resistant cases, systemic corticosteroids (e.g., prednisolone 1 to 2 mg/kg/d PO q.d.), then tapered over the next few months
For more resistant cases, systemic corticosteroids (e.g., prednisolone 1 to 2 mg/kg/d PO q.d.), then tapered over the next few months
 Necrotizing scleritis
Necrotizing scleritis
 Consider oral NSAIDs for 1 week.
Consider oral NSAIDs for 1 week.
 If there is no improvement, systemic corticosteroid can be added as above.
If there is no improvement, systemic corticosteroid can be added as above.
 For severe disease, oral corticosteroids can be initiated as above, or the patient can be hospitalized and treated with IV corticosteroids.
For severe disease, oral corticosteroids can be initiated as above, or the patient can be hospitalized and treated with IV corticosteroids.
 In resistant cases, other immunosuppressive therapy may be indicated (e.g., methotrexate, azathioprine, mycophenolate mofetil, cyclosporine, and cyclophosphamide). Biologics may be helpful in recalcitrant disease (e.g., infliximab, adalimumab, or rituximab).
In resistant cases, other immunosuppressive therapy may be indicated (e.g., methotrexate, azathioprine, mycophenolate mofetil, cyclosporine, and cyclophosphamide). Biologics may be helpful in recalcitrant disease (e.g., infliximab, adalimumab, or rituximab).
 Scleral (or pericardial or dural) patch graft may be required if there is a risk of perforation.
Scleral (or pericardial or dural) patch graft may be required if there is a risk of perforation.
 Topical corticosteroids are not effective
Topical corticosteroids are not effective
for scleritis. Topical cyclosporine 2% drops,
4 to 6 drops per day, may be of limited benefit.
 Subtenon’s corticosteroid injections are occasionally used, but they may cause scleral thinning and may increase the risk of perforation.
Subtenon’s corticosteroid injections are occasionally used, but they may cause scleral thinning and may increase the risk of perforation.
Prognosis
 Good for nonnecrotizing scleritis, guarded for necrotizing scleritis. The prognosis depends greatly on whether the underlying systemic disease can be identified and treated adequately.
Good for nonnecrotizing scleritis, guarded for necrotizing scleritis. The prognosis depends greatly on whether the underlying systemic disease can be identified and treated adequately.
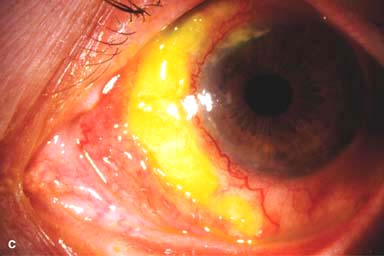
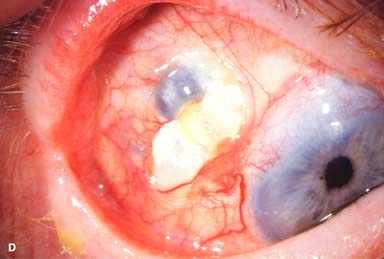
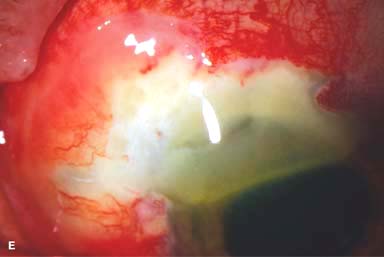
Figure 9-2. Scleritis. A. Diffuse inflammation of the conjunctiva and sclera is present in this eye with diffuse scleritis. The vessels did not blanch with topical phenylephrine 2.5%. B. An elevated, inflamed nodule of sclera can be seen. A purplish hue is present deep to the prominent vessels, suggesting scleral inflammation. Necrotizing scleritis. C. A large area of necrotizing scleritis with peripheral corneal involvement can be seen in this patient, who was eventually diagnosed with Wegener’s granulomatosis. Multiple scleral patch grafts, amniotic membrane grafts, and treatment with numerous systemic immunosuppressive agents failed to reverse the progression of this condition, which ultimately resulted in enucleation. D. An exposed scleral buckle has resulted in scleral melting with uveal show. It was treated with removal of the encircling band and a scleral patch graft. Corneoscleral melt. E. Ten days after limbal cataract surgery treated postoperatively with topical corticosteroids and generic diclofenac, a severe corneoscleral melt developed. Cultures were negative, and the condition did not improve with intensive topical fortified antibiotics. Two days later it perforated, requiring a large corneoscleral graft (see Chapter 10, Fig. 10-11B). F. A large area of patchy blue-gray discoloration of the superior sclera has resulted from scleral thinning from previous episodes of scleritis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


