Acute Proptosis in Childhood
Gerald J. Harris
Grant W. Su
The abrupt onset of proptosis in a child is an alarming event for the family and the physicians involved. The clinical situation evokes the specter of rhabdomyosarcoma and other malignant conditions in which prompt diagnosis and early therapy can be lifesaving. Other more common causes are not lethal but may cause vision loss. Still others are relatively benign in every sense.
The full differential diagnosis of pediatric proptosis is a broad one, although the causes are relatively distinct from those in adults (Table 27.1). The list of conditions that may produce sudden or rapidly progressive proptosis is considerably shorter, but a diverse group of disorders must still be considered in this clinical setting (Table 27.2). Some of these conditions originate in and are restricted to the orbit. They may result from disordered embryogenesis and development, or they may arise as primary neoplasms. In the pediatric population, tumors that metastasize from distant primary sites preferentially lodge within the orbit rather than within the globe, a reversal of the tendency noted in adults. Proptosis also may result from orbital involvement by systemic conditions, such as leukemia and the histiocytoses. Orbital manifestations of these disorders may be the presenting clinical signs. Finally, orbital inflammatory conditions can simulate true neoplasia and may have to be considered in the differential diagnosis in individual cases.
TABLE 27.1 Orbital Tumors/Lesions in Childhood | |
|---|---|
|
TABLE 27.2 Rapidly Expanding Orbital Masses in Children | ||
|---|---|---|
|
The general management of these disorders varies widely. In most of the malignant conditions, a prompt tissue diagnosis is followed by systemic evaluation and definitive multimodal therapy by a team skilled in pediatric oncology. In these cases, the preferred surgical approach for the biopsy may vary with the suspected tumor. For a lymphangioma with recent hemorrhage, the diagnosis might be established on clinical grounds and the patient observed, or intervention might be restricted to simple evacuation of blood cysts without attempts at major resection. For a dermoid cyst, complete excision without violation of the tumor capsule is the goal. For a capillary hemangioma, the clinical diagnosis generally is followed by simple observation without surgical intervention. In inflammatory processes, the diagnosis also is based on clinical evidence, and anti-inflammatory measures are initiated. Distinctions between infectious and idiopathic inflammations are necessary in choosing the appropriate medications.
Although these conditions are addressed individually elsewhere in this text, they are presented briefly and in juxtaposition here, because it is apparent that some informed distinctions must be made promptly at the time of initial presentation. Although we briefly discuss treatment, the emphasis of this chapter is on the features that might lead to a provisional diagnosis.
There are marked differences in the frequency of these disorders,1 but they do not reflect the clinical importance of any individual lesion. Statistical frequencies are also of less value in the diagnosis of any individual case than are careful analyses of the history, physical findings, imaging tests, and histopathologic samples. Computed tomography (CT) remains the first-line diagnostic test in most cases of proptosis. The topographic features shown by CT should narrow the differential diagnosis. Magnetic resonance imaging (MRI) can provide additional diagnostic information by exploiting different physical properties of the lesion compared with CT. For example, lesions that are isodense with normal structures by CT may not be isointense by MRI. In selected cases, orbital echography may further reduce the diagnostic choices by providing information about the lesion’s internal architecture.
Rhabdomyosarcoma
Rhabdomyosarcoma is the most common soft-tissue sarcoma in patients younger than 15 years of age and the most common primary orbital malignancy in childhood. These facts should not imply its frequent occurrence. Including all body sites, the annual incidence of childhood rhabdomyosarcoma in the United States is approximately 350 cases.2 The orbit is the site of origin in 5% to 25% of cases.3,4 However, site distribution varies with age. In children 5 to 9 years of age, for example, approximately 40% of primary rhabdomyosarcomas involve the orbit or eyelid.5 Although relatively rare, the tumor has a devastating natural history and demands a high index of suspicion in all cases of pediatric proptosis.
Orbital rhabdomyosarcomas are slightly more common in females, with a 0.79 to 1 male-to-female ratio.5 The average age of presentation is 7.8 years, but the tumor may be present at birth and has been reported in patients as old as 78 years.6 A positive family history and associated anomalies have at times been identified, but these are exceptions to the rule. Classically, orbital rhabdomyosarcoma presents in an abrupt manner, with rapid progression of proptosis over days to weeks. A somewhat more indolent course does not exclude the diagnosis, however. Vigilance also should be exercised when rapidly expanding eyelid lesions are encountered. Rhabdomyosarcoma may present as ptosis or an eyelid mass rather than with proptosis.4 An eyelid rhabdomyosarcoma can occur as a congenital lesion.7
Within the orbit, rhabdomyosarcoma occurs most often, but not exclusively, in the superior nasal quadrant, with downward and outward displacement of the globe. CT scans show the topography of the orbital mass (Fig. 27.1A), as well as the possible extension into adjacent bone, paranasal sinuses, or the intracranial cavity. The circumscription that may be noted on CT is relative, because the lesion is not encapsulated and microscopically infiltrates normal tissue. Echography shows internal echoes of low-to-medium amplitude. Because the cellular tumor absorbs acoustic energy, the amplitude of the spikes falls off somewhat through the lesion (Figs. 27.1B and 1C), MRI can help define the tumor’s relationship to extraocular muscles (Fig. 27.2).
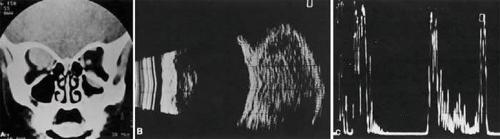 FIGURE 27.1 A: Proptosis and downward, outward globe displacement developed over 2 days in a 3-year-old girl. A homogeneous mass fills the superomedial orbit. B: Contact B-scanning shows a relatively well-circumscribed mass with uniform internal echoes. C: Contact A-scanning shows the internal reflectivity to be of low-to-medium amplitude, consistent with a sarcomatous lesion. Biopsy results confirmed the diagnosis of rhabdomyosarcoma. |
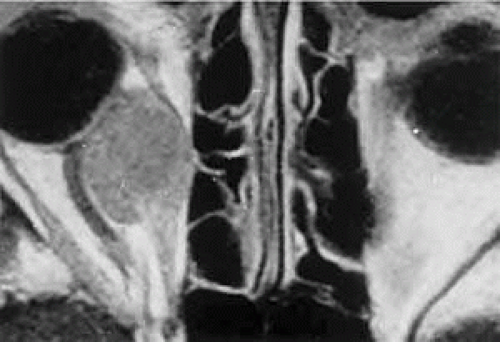 FIGURE 27.2 MRI shows an intraconal tumor of lower intensity than the medial rectus muscle. The proximal muscle is splayed rather than compressed, suggesting that the lesion originated within the medial rectus. The diagnosis was alveolar rhabdomyosarcoma. |
The clinical diagnosis must be confirmed by biopsy. Because of the risk of seeding the biopsy tract, a transcranial approach should be avoided. If possible, the periosteum should not be violated because it presents a relative barrier to tumor invasion. Depending on its location, the lesion should be approached transconjunctivally or with an eyelid crease incision and transeptal dissection. The surgeon must balance the benefit of complete gross tumor resection with the risks of functional impairment and tumor dissemination that may accompany that effort. Tissue samples should be fixed in formaldehyde solution and glutaraldehyde for light and electron microscopic study, respectively. In addition, the value of immunohistochemical differentiation has been established for some time, and the potential uses of molecular genetic studies are rapidly emerging. Consequently, the procurement of fresh or frozen tissue, or both, has been given the highest priority by the Biopathology Discipline within the Intergroup Rhabdomyosarcoma Study Group (IRSG).5 These techniques can facilitate the diagnosis of poorly differentiated tumors, and they may refine diagnostic and prognostic classifications, identify candidate genes, and contribute to potential gene therapies.8,9
Since the inception of IRSG-I in 1972, the multicenter collaboration has enrolled the overwhelming majority of patients diagnosed with rhabdomyosarcoma in the United States and has contributed significantly to enhanced patient survival. Patients with orbital tumors had a 96% versus 83% failure-free survival in IRSG-IV compared with those in the IRSG-III.5,10 As of 2005, the overall (all primary sites) 5-year survival of children and adolescents with nonmetastatic and metastatic tumors approached 70%.11 This progress reflects advances in diagnostic imaging and multimodal treatment, including chemotherapy (e.g., agents, combinations, timing), radiation therapy (e.g., doses, fractionation, timing), and surgery (e.g., diagnostic biopsy, local staging, salvage procedures).
Therapeutic protocols have evolved over the past 30 years, but they also have not been uniform at any given point in time. Rather, they have been tailored to the patient’s level of risk, as determined by multiple prognostic factors (Table 27.3). The concept of “risk-appropriate therapy”12 recognizes, for example, that a 6-year-old child with an embryonal rhabdomyosarcoma confined to the orbit might do well with a relatively simple chemotherapy protocol, avoiding the late adverse effects of high-dose radiation. Conversely, an 18-year-old patient with an alveolar rhabdomyosarcoma arising in the retroperitoneum, with metastases at presentation, needs aggressive, complex chemotherapy and radiation, and may still do poorly. Prognostic factors considered by the multidisciplinary team include: the presence of gross or microscopic residual tumor, and this determination currently is being redefined with molecular techniques that may show residual disease even without microscopic evidence;5 whether tumor is confined to the anatomic site of origin or invades surrounding tissues; tumor size, with 5 cm considered a breakpoint; regional lymph node involvement; and distant metastasis. Body site plays a role, and the orbit is relatively favored. The age of the patient at diagnosis is a strong independent predictor of outcome.12 The current pathologic classification for childhood rhabdomyosarcomas by prognosis5 is as follows:
Superior prognosis: botryoid, spindle cell
Intermediate prognosis: embryonal
Poor prognosis: alveolar, undifferentiated, anaplastic (formerly pleomorphic)
Indeterminate prognosis: rhabdomyosarcoma with rhabdoid features
TABLE 27.3 Prognostic Factors in Rhabdomyosarcoma | |
|---|---|
|
Although no single regimen is appropriate for every child with orbital rhabdomyosarcoma, a sample protocol might include multiple 3-week cycles of chemotherapy, each beginning with intravenous vincristine, actinomycin-D, and cyclophosphamide, with vincristine repeated on the 8th and 15th days of each cycle. The regimen might include external radiation to a total dosage of 5,040 cGy. For poor prognosis cases (e.g., metastatic alveolar rhabdomyosarcoma), newer agents under investigation include ifosfamide, etoposide, irinotecan, topotecan, and tirapazamine.5,11
Having made the diagnosis and contributed to local staging at the time of presentation, the orbital surgeon continues to follow the patient along with the pediatric oncology team. In cases of treatment failure, “salvage” surgery may take the form of orbital exenteration13 or excision of residual tumor combined with brachytherapy.14
Rhabdomyosarcoma underscores the importance of clinical suspicion when dealing with acute proptosis in childhood. Prompt referral to a tertiary center after appropriate imaging is the responsibility of the primary ophthalmologist, family practitioner, or pediatrician who first encounters the patient.
Other Primary Sarcomas
Other primary orbital sarcomas may have a relatively abrupt onset of proptosis, although their progression generally is less explosive than that of rhabdomyosarcoma. As with rhabdomyosarcoma, a prompt biopsy is critical for appropriate management.
Alveolar soft part sarcoma, an example of this group of lesions, is a rare tumor that may affect the pediatric orbit. In an extensive review of the literature, Sullivan, Mortimore, and Fulcher15 identified about 50 orbital cases. They noted that the tumor tends to involve the extremities of young adults or the head and neck region of children, with a predilection for the orbit and tongue. A myogenic origin is favored, but there also is evidence for a neural derivation. The findings of imaging studies are nonspecific. Diagnosis depends on the light and electron microscopic demonstration of periodic acid–Schiff-positive, diastase-resistant crystals within the cytoplasm of large polyhedral tumor cells.16 Pediatric patients appear to have a better prognosis than do adults. The currently recommended treatment is local excision of circumscribed primary lesions, with exenteration reserved for diffuse orbital involvement or local recurrence. Radiation therapy and chemotherapy may have adjunctive value.
Epithelioid sarcoma is a rare, high-grade soft tissue tumor that can occur in older children and young adults. Most lesions originate in the distal upper extremities. Epithelioid sarcoma has a local recurrence rate of up to 69% and a rate of regional lymph node metastasis of up to 44%, but an overall 5-year survival of up to 66%.17 White et al.18 identified two patients with orbital lesions; one was a 17-year-old girl with primary epithelioid sarcoma of the orbit. The tumors have both mesenchymal and epithelial histologic qualities and grow in tendon sheaths in a grossly nodular pattern. Treatment strategies include wide local excision and adjuvant radiotherapy, as well as chemotherapy for metastatic disease.19
Neuroblastoma
Neuroblastoma is the most common metastatic orbital lesion in children.20 It represents 10% to 15% of all pediatric malignancies, ranking behind only leukemia and solid central nervous system (CNS) tumors in frequency. It is a tumor of primitive neuroblastic tissue and, in some respects, is the autonomic nervous system counterpart of retinoblastoma. It usually originates in the adrenal medulla or other retroperitoneal sites, but also may arise from any of the sympathetic ganglia in the mediastinum or neck. Neuroblastoma typically afflicts children from 18 months to 3 years of age, although it may be present at birth or may not appear until the mid-teens.
In a review of more than 400 cases of neuroblastoma, Musarella et al.21 found the incidence of ophthalmologic signs to be 20%. In almost half of these cases, the ocular symptoms were the presenting complaints. The most common eye signs were related to orbital metastasis. Orbital involvement was bilateral in approximately half of the cases. Characteristic eye findings include proptosis and periorbital ecchymosis. The latter results from hemorrhagic necrosis within a rapidly growing tumor that has outstripped its blood supply. Other eye signs may reflect more distant tumor involvement. Horner syndrome can result from a primary neuroblastoma in the sympathetic ganglia of the neck or mediastinum or from metastases to either of these regions.20,21,22 An infantile Horner syndrome is characterized by hypochromia of the ipsilateral iris. Ocular signs may include opsoclonus, a wild conjugate oscillation of both eyes that may be associated with myoclonus, and truncal ataxia.21 It has been proposed that this complex results from an antibody directed against neuroblastoma antigen, which may cross-react with cerebellar tissue, producing damage in that area.23 Neuroblastomas manufacture catecholamines, which, in sufficient quantity, can produce flushing, systemic hypertension, and diarrhea. Diagnosis is aided by the demonstration of elevated urinary catecholamine metabolites (e.g., homovanillic acid, vanillylmandelic acid) in the urine.24
In cases of suspected neuroblastoma metastatic to the orbit, the primary tumor may be shown by abdominal or thoracic imaging studies. A histologic diagnosis generally is required, however. Tissue may be obtained from bone marrow aspiration or biopsy of primary or secondary sites. Orbital soft tissue involvement usually follows extension from bony metastasis (Fig. 27.3). Therefore, orbital biopsies should be performed extraperiosteally, because the periorbita may still be intact and constitute a relative barrier to tumor extension. Because the histologic differential diagnosis includes other poorly differentiated tumors of childhood, specimens should be fixed in both formalin and glutaraldehyde, and fresh tissue also should be submitted.
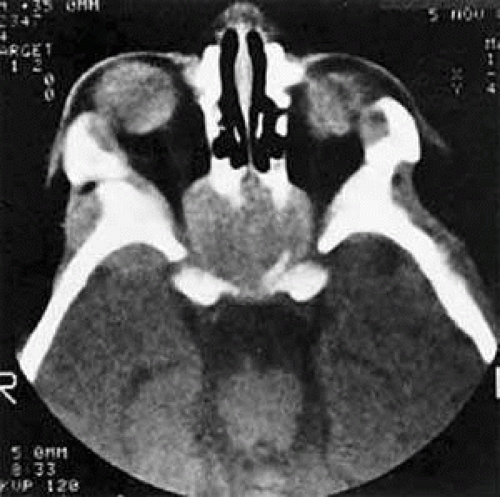 FIGURE 27.3 A large metastatic focus of neuroblastoma has destroyed the body of the sphenoid bone and has extended into both orbital apices. A second site involves the outer portion of the right sphenoid wing and extends into the orbit and the middle cranial and temporal fossas. The tumor originated in the right adrenal gland. |
Histologically, neuroblastomas display features commensurate with their degree of differentiation. At the more primitive end of the spectrum are tumors comprising small round cells with minimal cytoplasm. At the other extreme are lesions consisting of large, cytoplasm-rich elements resembling ganglion cells. It has been proposed that neuroblastomas having undergone spontaneous clinical regression have evolved into benign ganglioneuromas.25 Homer-Wright pseudorosettes characteristically present in well-differentiated primary neuroblastomas and are rarely, if ever, found within orbital metastases. In poorly differentiated tumors, electron microscopy may be required to show neurosecretory granules containing catecholamines. The rapidly advancing field of immunohistochemistry also has contributed to diagnosis and prognostic assessment in neuroblastoma.26
As with rhabdomyosarcoma, the choice among treatment protocols for neuroblastoma is based on tumor staging and the multiple prognostic factors that have been identified in large cooperative trials. Approximately 25% of children with newly diagnosed neuroblastoma present with nonmetastatic and localized disease.26 This group has a 98% survival with surgery alone as primary therapy. However, children with localized disease who have amplification of the N-myc oncogene, or who are 2 years of age or older with either unfavorable histopathology or positive lymph nodes, are at greater risk of death. Other negative factors include elevated serum levels of ferritin and neuron-specific enolase.
The majority of children with neuroblastoma have metastatic disease at diagnosis.27 In order of frequency, the most common sites are bone marrow, bone, lymph nodes, liver, intracranial and orbital sites, lung, and central nervous system. The metastatic pattern differs with age. Patients younger than 1 year are more likely to have liver or skin metastases at diagnosis and less likely to have bone and bone marrow metastases at diagnosis than do patients age 1 year or older. Among children with metastases at diagnosis, event-free survival is decreased in patients with bone, bone marrow, central nervous system, intracranial/orbital, lung, and pleural metastases; and is improved in those with liver and skin metastases.
Depending on tumor staging and risk factors, treatment protocols may include surgical resection of the primary tumor, combination chemotherapy (cyclophosphamide, iphosphamide, vincristine, doxorubicin, cisplatin, carboplatin, etoposide, or melphalan) of varying dose-intensity, and myeloablative therapy with autologous purged bone marrow transplantation.26 In high-risk neuroblastomas, aggressive surgical treatment with local irradiation and myeloablative chemotherapy with stem cell rescue have been recently shown to provide good local control.28 In the future, gene expression profiling may allow physicians to tailor therapy for each patient, with improvement of clinical outcome and survival rates.29
Other Metastatic Lesions
Orbital metastases from other solid pediatric tumors are less common. Of these, Ewing sarcoma accounts for the majority, and Wilms tumor is responsible for an extremely small number of cases.20,30
Ewing sarcoma accounts for approximately 10% of tumors that metastasize to the pediatric orbit. Albert, Rubenstein, and Scheie20 noted orbital involvement in 5 of 12 patients with Ewing sarcoma. In all patients, the orbital metastases were unilateral and were clinically noted several months after diagnosis of the primary lesion. Ewing sarcoma usually arises within the medullary canals of the bones of the trunk or extremities. Unlike neuroblastoma, its peak incidence is in late childhood and adolescence. Ewing sarcoma and malignant peripheral neuroectodermal tumor (PNET) are closely related, but are distinguished by the microscopic and immunohistochemical findings of greater neuroectodermal differentiation (e.g., rosette formation) in the latter lesion.31,32 Although earlier studies suggested a poorer prognosis in PNET than in Ewing sarcoma, more recent work showed no difference in clinical outcome.33,34 Fluorine-18 fluorodeoxyglucose positron emission tomography (FDG-PET) has been recently reported to be highly sensitive in detecting bone metastases in Ewing sarcoma and in assessing the response to neoadjuvant chemotherapy.35 The principal adverse prognostic factor is metastasis at diagnosis. In a European study of 975 patients enrolled from 1977 to 1993, the 5-year relapse-free survival of patients without metastases at diagnosis was 55%, compared to 22% for patients with metastases at diagnosis.36 During the second 8 years of the study, these figures were 60% and 30%, respectively, indicating continued outcome improvement. Although most cases of Ewing sarcoma in the orbit represent metastatic disease, several primary orbital tumors have also been reported.37,38
Wilms tumor (nephroblastoma) arises from embryonic elements within the kidney. Although it affects children almost as frequently as neuroblastoma and can metastasize extensively to other sites, orbital lesions have been rarely described.30,39 Reports have concerned children younger than 3 years of age. Multimodality treatment with chemotherapy, surgical resection, and radiation therapy has yielded overall survival rates of 90%.40 Most patients have favorable histology (nonanaplastic or focally anaplastic tumors) and survive after preoperative chemotherapy and nephrectomy.41,42,43 However, poor outcomes are associated with diffuse anaplasia, chromosomal loss on 1p and 16q, diploidy, lung or liver metastases, major tumor spillage during resection, remote lymph node involvement, and bilateral tumors.
Acute Leukemia
Leukemia is the most common malignancy in childhood, and nearly all pediatric leukemias are acute rather than chronic. The lymphoblastic variety is approximately four times more common than the myelogenous form. Leukemic cells frequently lodge in the eye and adnexa, and bilateral involvement is common.44 Proptosis occurs less often than intraocular or optic nerve complications and may result from a combination of local tumefaction and hemorrhage. Most ophthalmic complications are associated with acute lymphoblastic leukemia (ALL) rather than with acute myelocytic leukemia (AML). Orbital infiltration may occur in either condition but is disproportionately more common in AML.45,46 Although prior studies47,48,49 have reported that most orbital leukemic tumors precede the diagnosis of systemic disease, a recent study by Bidar et al.50 demonstrated that the majority of patients (23 out of 27) were diagnosed with systemic leukemia before manifestation of the orbital disease. Nevertheless, the orbital disease was detected during the systemic workup in 10 of the 23 patients during the first 2 weeks following the initial diagnosis. Therefore, although orbital disease may occur at any time during the course of leukemia, it tends to occur early, and ophthalmologists may be the first to diagnose this systemic disease.
Extramedullary deposits of primitive myeloblasts in AML have been termed chloromas, because of a greenish hue imparted by the enzyme myeloperoxidase within the tumor cells. This discoloration fades on exposure to the air and is therefore an inconstant finding at the time of histopathologic preparation and diagnosis.51 Granulocytic or myeloid sarcoma is considered a more appropriate term and is preferred. Favored sites of involvement are the bones and periosteum of the skull, including those of the orbit. Granulocytic sarcomas also may occur in the eyelids as well as the orbital soft tissues, including the lacrimal gland and extraocular muscles.52,53
Granulocytic sarcomas of the orbit may be bilateral in 10% to 45% of cases.47,48 Hemorrhage occurs frequently, and eyelid ecchymosis may be a presenting sign. Most patients are affected in the first decade of life, and males are involved more often than females. The majority of cases in the older series cited were derived from Asia, Africa, or the South Pacific, and, interestingly, granulocytic sarcoma was the second most common cause of proptosis in Uganda after Burkitt lymphoma.
Findings in imaging studies typically reveal homogenous masses that mold to orbital walls without bone destruction (Fig. 27.4).50 With nonenhanced CT, orbital masses appear as isodense or slightly hyperattenuated relative to brain and muscle. With intravenous contrast infusion, CT shows mild homogenous enhancement. On T1-weighted MRI, orbital masses are usually isointense to gray matter and muscle. Gadolinium-enhanced T1-weighted MRI shows mild enhancement relative to gray matter and muscle. On T2-weighted MRI, masses are typically isointense to white matter and muscle.
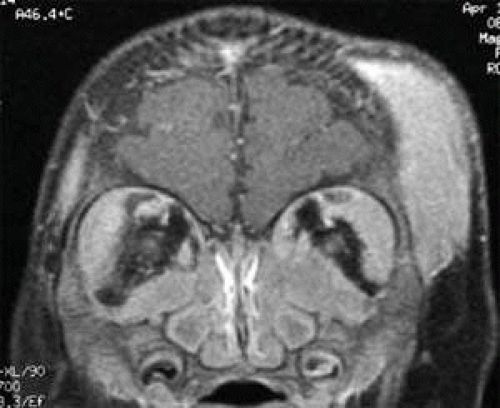 FIGURE 27.4 Granulocytic sarcoma involving both orbits and the left temporal scalp. |
Given the advances in imaging and diagnostic evaluation of leukemic tumors, a confirmatory orbital biopsy may not be needed. However, if a diagnosis of systemic leukemia has not been previously established, orbital biopsy should be performed. As in other pediatric tumors, light microscopy may yield ambiguous findings, and immunohistochemical stains and electron microscopy are helpful. Granulocytic sarcomas are composed of large mononuclear cells that resemble histiocytes. Diagnosis may be difficult when immature myeloblasts dominate the histologic picture and evidence of granulocytic differentiation is minimal. Diagnosis is aided in these cases by the Leder stain, which indicates esterase activity and cellular differentiation in a myelocytic direction. In addition, the immunohistochemical stain for lysozyme is positive in 60% to 89% of cases. In patients in whom both the Leder and the lysozyme stains are negative, the monoclonal antibody MAC387 may establish the diagnosis.49 Electron microscopy may show early granule formation.
Due to improvements in diagnostic imaging and treatment, the survival rate for AML improved from 13% in the 1970s to 40% in the 1990s.54 During that same interval, survival for ALL increased from 53% to 81%. In a recent study of orbital leukemic tumors, Bidar et al.50 reported a survival rate of 55% (11/21) for AML patients and 60% (3/5) for ALL patients. Prognosis improves with bone marrow transplants from histocompatible sibling donors early in the first remission.55,56 Molecular genetic advances are expected to improve therapeutic strategies.
Burkitt Lymphoma
Although it is a rare cause of proptosis in the Western hemisphere, Burkitt lymphoma deserves attention because of its distinctive epidemiologic and clinical features. The tumor occurs endemically within certain geographic and climatic boundaries in East Africa. It is the most common pediatric orbital tumor in Uganda, accounting for almost 50% of cases.57 The average age of presentation is 7 years, with a range of 3 to 15 years. Large extranodal tumors occur in the bones of the jaw and the abdominal viscera. Unilateral or bilateral proptosis is present in 20% of cases and usually results from maxillary extension. The progression of proptosis may be explosive. Burkitt lymphoma has a doubling time that may be as brief as 24 hours, ranking it as the fastest growing tumor in humans.58 Endemic African cases have been linked to the Epstein-Barr virus and to a t(8;14q) chromosomal translocation, suggesting an interaction between environmental factors and host susceptibility.
In the United States and western Europe, Burkitt lymphoma accounts for up to 40% of lymphoma in children (and 1%–2% of lymphoma in adults).59 Sporadic North American cases have a less definitive viral association than endemic African cases. These patients also differ clinically in their age of presentation (mean, 11 years) and in the usual site of tumor origin (intra-abdominal lymphoid tissue).60,61 Involvement of the facial bones and orbit is less common in the North American cases, but invasion of the orbit from the sinuses may occur (Fig. 27.5).62,63
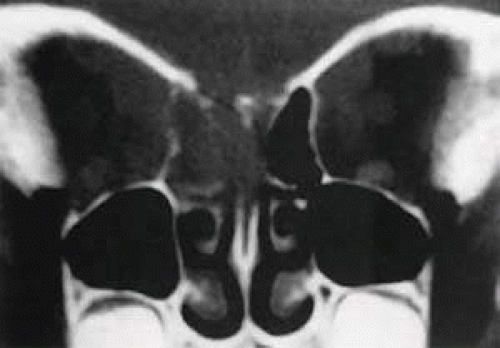 FIGURE 27.5 Burkitt lymphoma involving the posterior ethmoids, skull base, and both orbital apices in a 5-year-old boy. |
Biopsy is necessary to establish the diagnosis. The characteristic microscopic picture is that of a “starry sky,” made up of a homogeneous background of neoplastic lymphocytes and interspersed larger histiocytes with more abundant cytoplasm.
Burkitt lymphoma was considered a small noncleaved cell lymphoma in the Working Formulation and was identified as a peripheral B-cell neoplasm in the Revised European-American Lymphoma Classification. Burkitt lymphoma subsequently was subdivided into endemic, sporadic, immunodeficiency-associated, and atypical forms in the World Health Organization scheme.64 In children, survival rates of 80% are being achieved with intensive, short-duration chemotherapeutic protocols.65 Features associated with adverse outcomes include older age, advanced stage, poor performance status, bulky disease, high lactate dehydrogenase, and CNS or marrow involvement.66
Langerhans-Cell Histiocytosis
Unifocal Langerhans-cell histiocytosis (LCH) or eosinophilic granuloma is a relatively benign, probably reactive lesion composed of histiocyte-type cells and an inflammatory infiltrate of eosinophils and neutrophils.67 Of the disorders traditionally grouped under the histiocytosis X rubric—acute disseminated LCH or Letterer-Siwe disease, multifocal LCH or Hand-Schüller-Christian disease, and unifocal LCH—the last is the most frequent cause of orbital disease. It generally affects the superotemporal quadrant as an extension from an osteolytic lesion (Fig. 27.6).67,68,69 There is a male predominance with onset in the first or second decade. Symptoms include bone pain, tenderness, and local swelling. The differential diagnosis, based on initial CT studies, includes other primary causes of bone erosion in this region, such as dermoid cysts and lacrimal neoplasms, as well as tumors metastatic to orbital bone, such as Ewing sarcoma. Diagnosis ultimately requires microscopic analysis of tissue (Fig. 27.7). Percutaneous fine-needle aspiration, with or without core-needle biopsy, has been used for lesions of the extremities70 and can be considered for orbital lesions. However, the need for general anesthesia in children, the risk of uncontrollable hemorrhage, and the frequent extension of tumor to the dura all weigh toward open biopsy. Expression of CD1a, detected by immunohistochemistry, is considered diagnostic of LCH (Fig. 27.7C).67 Electron microscopy shows “Langerhans” or “Birbeck” granules, with a characteristic tennis-racket shape (Fig. 27.7D).
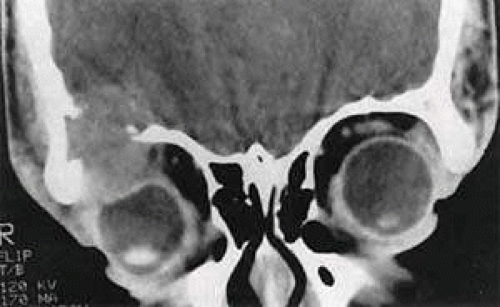 FIGURE 27.6 Langerhans-cell histiocytosis in a 16-year-old boy. The growth pattern suggests an intraosseous origin. Source: Harris GJ, Beatty RL: Acute proptosis in childhood. In Linberg JV (ed): Oculoplastic and Orbital Emergencies. Norwalk, CT: Appleton & Lange, 1989:93. |
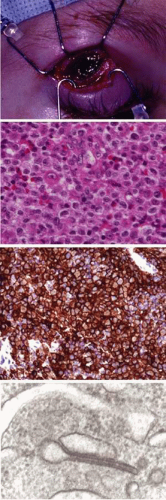 FIGURE 27.7 Langerhans-cell histiocytosis. A: Exposure through eyelid crease incision of a relatively large, hemorrhagic, gelatinous orbital component. B: Dense infiltrate of Langerhans-type histiocytes and eosinophils, with scattered lymphocytes. Pathologic Langerhans cells are large, mononuclear, and polyhedral, with abundant pink cytoplasm and ill-defined borders; nuclei are large, oval, and longitudinally grooved or clefted, with somewhat vesicular chromatin and occasionally distinctive nucleoli (hematoxylin and eosin, original magnification ×400). C: Immunohistochemical staining strongly confirms expression of the CD1a antigen by the Langerhans-type cells (original magnification ×200). D: Electron microscopic demonstration of intracytoplasmic Birbeck (Langerhans) granules. Some have a characteristic tennis-racket configuration (original magnification ×96,000). Source: Reprinted from Woo KI and Harris GJ. Eosinophilic granuloma of the orbit: Understanding the paradox of aggressive destruction responsive to minimal intervention. Ophthal Plast Reconstr Surg 19:429–439, 2003, with permission. |
Local treatment options have included surgical curettage, low-dose irradiation (400 to 800 cGy), and intralesional steroids. CT-guided steroid injection48 offers a safer alternative than blind injection, but some of the concerns about diagnostic needle aspiration still apply. Interestingly, despite significant bone destruction, complete resolution after diagnostic biopsy or relatively minor local intervention has been well documented (Fig. 27.8).67,71
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


