Chapter 49 Acquired and other retinal diseases (including juvenile X-linked retinoschisis)
Diabetic retinopathy
Diabetic retinopathy, while uncommon in children, is strongly correlated with duration of diabetes and overall glycemic control (percentage of glycosylated hemoglobin). The prevalence of diabetic retinopathy rises steadily following puberty, with a 4.8 times greater risk of postpubescent adolescents developing retinopathy relative to pubescent or prepubescent children with the same duration of diabetes.1 The progression of diabetic retinopathy associated with puberty is a combination of metabolic changes as well as decreased patient compliance.
Until recently, pediatric diabetes was almost exclusively the type 1 insulin-dependent form. With increasing obesity in the pediatric population, however, type 2 diabetes and insulin resistance is an emerging epidemic in children and adolescents, comprising 8–45% of all juvenile diabetes.2 Initial screening of diabetic children by an ophthalmologist 3–5 years following diagnosis remains critical; in a cohort aged 13–17 years intensive insulin therapy delayed the onset and reduced the progression of diabetic retinopathy.3
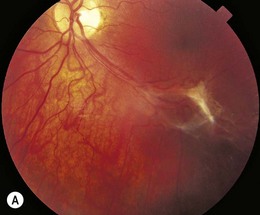
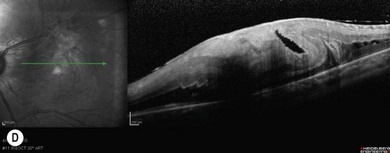
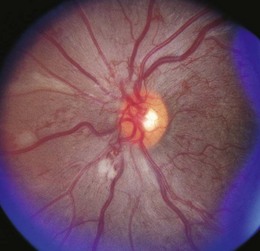
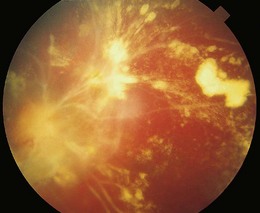
Retinopathy is slowly progressive, affecting between 5% and 44% of insulin-dependent diabetic children after 5 years of systemic disease and nearly 100% within 20 years.4–6 Longitudinal studies demonstrate clinically significant macular edema after 7 years of systemic disease, with a linear yearly cumulative risk as high as 6.7% between 10 and 20 years of duration.7 Proliferative diabetic retinopathy (Fig. 49.1), though extremely rare in the pediatric population, has been observed in children as young as 13 years old with an 8-year history of diabetes.8
Sickle cell retinopathy
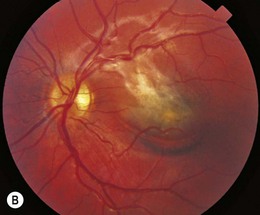
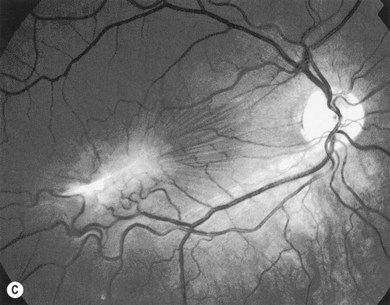
Ocular manifestations of sickle cell (SC) disease are most commonly seen in hemoglobin SC disease, homozygous SC disease, SC thalassemia, and heterozygous SC trait. Under conditions of hypoxia, dehydration, acidosis, and hyperviscosity, the abnormal hemoglobins polymerize, resulting in sickled erythrocytes.9 Sickled erythrocytes are less pliable and cannot easily migrate through small capillaries or pores of the trabecular meshwork. Retinal non-perfusion initiates a cascade of vascular remodeling at the junction of perfused and non-perfused retina. Arteriovenous anastomoses may develop, eventually giving rise to “sea fan” proliferative retinopathy (Fig. 49.2), most commonly seen in SC and SC thalassemia disease.10–12 Intraretinal salmon patch hemorrhages are often observed, which upon resorption may result in refractile spots of hemosiderin deposition beneath the internal limiting membrane. Black “sunburst” lesions, the sequela of subretinal hemorrhage, are focal areas of retinal pigment epithelial hypertrophy and hyperplasia.13
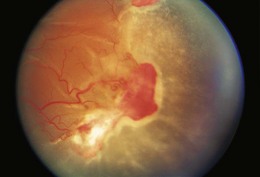
Proliferative SC retinopathy may lead to vitreous hemorrhage, tractional and rhegmatogenous retinal detachments, but more frequently undergoes spontaneous regression.14,15 Suggested mechanisms for autoinfarction include feeder arteriolar occlusion, capillary occlusion, or hemodynamic alterations induced by vitreous traction.16,17 Peripheral scatter photocoagulation may induce regression and reduce the risk of vitreous hemorrhage.18 In the event of retinal detachment, caution should be taken to minimize the risk of anterior segment ischemia as reported with encircling scleral buckles.19
Traumatic hyphema in children with SC hemoglobinopathies poses an increased risk of complications, including raised intraocular pressure as a result of aqueous outflow obstruction by sickled erythrocytes.20,21 Even mild elevations in intraocular pressure may produce sludging in the vascular supply to the optic nerve and retina, leading to central retinal artery occlusion or optic atrophy.22 Therefore, surgical intervention or antifibrinolytic therapy with aminocaproic acid may be necessary. Additionally, there is an increased risk of rebleeding after the initial hyphema in pediatric SC patients.23
Radiation retinopathy
Radiation retinopathy24,25 is seen in children secondary to focal plaque (brachytherapy) and external beam treatments for retinoblastoma and orbital tumors.26,27 The total dose and the fraction size of radiation are the key elements predisposing to radiation retinopathy, with retinal vascular damage occurring at lower doses with external beam than with plaque therapy.26 The threshold for retinopathy is a total dose of 3000–3500 cGy with external beam radiotherapy, but retinopathy has been reported with as little as 1500 total cGy.26,28 Radiation retinopathy most commonly develops between 6 months and 3 years following treatment; retinal changes have been observed as early as 1 month following both plaque and external beam.29–31
Radiation induces a cascade of changes in the retinal vascular architecture, predominantly in the macula, which lead to vascular non-perfusion, incompetence, and proliferation. Histologic studies demonstrate a preferential loss of vascular endothelial cells with a relative sparing of the pericytes, a distinct contrast to the early loss of pericytes in diabetic retinopathy.29 Progressive endothelial cell loss results in capillary occlusion and irregular dilatations, telangiectasias, and microaneurysms of the neighboring capillary bed.32 More severe and extensive radiation retinopathy is associated with diabetes mellitus and the administration of chemotherapeutic agents.26,33,34
Bone marrow transplant retinopathy
An ischemic retinopathy, similar to that observed with radiotherapy, is seen following bone marrow transplantation (BMT) for hematologic malignancies and aplastic anemia. The retinopathy is decreasing in prevalence as better chemotherapy makes BMT less necessary and stem cell transplants reduce the immunologic complications. The retinal manifestations include capillary non-perfusion, cotton-wool spots, and intraretinal hemorrhages (Fig. 49.3).35 The vasculopathy generally develops within 6 months of BMT, with histologic changes resembling those of early radiation retinopathy.35,36 While the majority of ocular complications associated with BMT are cataract and dry eye, the incidence of ischemic retinopathy and disc swelling ranges from 6.9% to 13.5%.35,37–40 In a study of children undergoing bone marrow transplantation, 95.7% of eyes retained visual acuity of 20/40 or better, with poor visual outcomes associated with cytomegalovirus retinitis, a presumed submacular Nocardia abscess, corneal ulcers secondary to dry eye syndrome, and cataracts.40
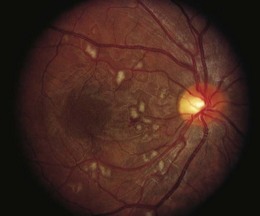
The pathogenesis of the retinopathy in BMT patients remains unclear, confounded by the conditioning regimens prior to transplantation which typically involve total body radiation and chemotherapy, as well as the administration of agents following transplantation designed to suppress graft-versus-host disease. The chemotherapeutic agents may be directly toxic to the retinal vasculature, potentially increasing the susceptibility to retinopathy, as retinopathy develops with relatively low doses of total radiation40,41 as well as in its absence.42
Retinal vasculitis
Retinal vasculitis may occur secondary to a systemic disease or an infectious agent, or as an isolated retinal etiology. Posterior segment manifestations include vascular sheathing, intraretinal hemorrhages, retinal exudates, vitritis, as well as macular edema. Both vascular incompetence and ischemia may be seen on fluorescein angiography; vessel staining or leakage are common findings. Non-perfusion may produce cotton-wool spots (Fig. 49.4), intraretinal whitening, and proliferative retinopathy. Fundus features are often non-specific, and other ocular or systemic features may aid in the diagnosis of the underlying etiology.
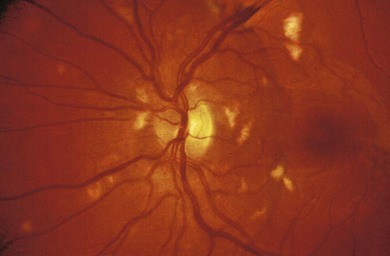
Fig. 49.4 Retinal vasculitis in a child with systemic lupus erythematosus. Shows multiple cotton-wool spots.
Causes of retinal vasculitis in the pediatric population include collagen vascular disorders,43 Behçet’s disease,44 Eales’ disease,45 acquired toxoplasmosis,46 multiple sclerosis,47 human immunodeficiency virus, human T-cell lymphotropic virus type-1,48 common variable immunodeficiency syndrome,49 Henoch-Schönlein purpura,50 and the syndrome of idiopathic vasculitis, aneurysms and neuroretinitis.51 Treatment is tailored to the underlying etiology and retinal pathology.
Frosted branch angiitis
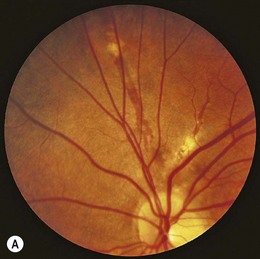
Frosted branch angiitis is a syndrome of acute, often bilateral, vision loss in otherwise healthy patients, commonly children and young adults.52 Fundus examination reveals thick inflammatory sheathing surrounding the veins (and arteries to a lesser extent), extending from the optic nerve to the periphery (Fig. 49.5). The visual acuity is often reduced to counting fingers, but may be relatively unaffected in mild cases.53 Intraretinal hemorrhages, serous macular detachment, optic disc swelling, conjunctival injection, anterior uveitis, and vitreous inflammation may be present.54,55 Fluorescein angiography demonstrates late staining and leakage from the veins without any evidence of stasis or occlusion.
Systemic corticosteroids remain the mainstay of treatment, with prompt resolution of perivascular infiltrates and recovery of vision in the majority of cases.54–57 A similar recovery with topical or subconjunctival steroids alone has been reported.53,57,58 Visual recovery, however, may be limited by macular scarring, branch retinal vein occlusions, and diffuse retinal fibrosis.54,59
The origin of frosted branch angiitis is uncertain, though the efficacy of corticosteroids suggests an underlying immune response. Frosted branch-like appearances have been associated with cytomegalovirus retinitis in immunosuppressed patients,60–63 and with autoimmune diseases such as systemic lupus erythematosus64 and Crohn’s disease.65
Angioid streaks
Angioid streaks are irregular linear streaks of variable pigmentation which may radiate in all directions from the peripapillary retina (Fig. 49.6). The streaks taper as they approach the optic disc, producing a circumferential peripapillary ring. The lesions are typically bilateral and vary in coloration depending on the pigmentation of the fundus. Angioid streaks are not seen at birth and only rarely have been observed in children, with earliest observations at 8 years of age.66,67 Angioid streaks have been noted to increase in length and width over time. The incidence of angioid streaks increases with age, especially in the sickle hemoglobinopathies in which angioid streaks are typically observed only after the age of 25 years.68,69
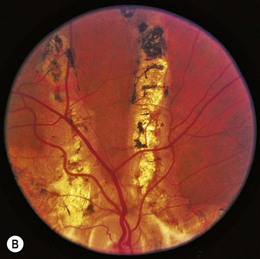
The term angioid streak was originally coined by Knapp, reflecting the prevailing notion that this fundus abnormality had a vascular etiology.70 Subsequent clinical and histopathologic studies have demonstrated that the streaks represent localized breaks at the level of Bruch’s membrane.80 Indocyanine green has proven to be a useful diagnostic tool in identifying angioid streaks and their associated ocular pathology.71
Angioid streaks, in up to 50% of cases, are associated with a systemic condition, most commonly pseudoxanthoma elasticum (PXE) (Fig. 49.7), Paget’s disease of bone, and sickle hemoglobinopathies.72 Extensive calcification of Bruch’s membrane has been demonstrated in cases of Paget’s disease and PXE, potentially rendering Bruch’s membrane more brittle and subject to breaks. In hereditary spherocytosis, iron deposition may predispose Bruch’s membrane to breaks.73 The distribution of angioid streaks may represent the effects of biomechanical forces and the traction exerted by the extraocular muscles.74
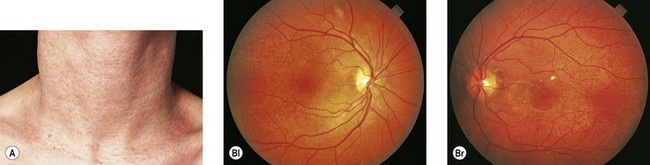
Choroidal neovascularization, though a rare phenomenon in children, may develop secondary to the breaks in Bruch’s membrane, resulting in serous detachments, subretinal hemorrhage, and disciform scarring. Fundus precursors to angioid streaks may be seen in children with PXE, and include a peau d’orange pigment speckling in the posterior pole (Fig. 49.7), as well as peripheral salmon spots, yellow deposits in the retinal pigment epithelium (RPE).66,67 Subretinal hemorrhages following blunt ocular trauma have been observed in eyes with angioid streaks.75 Identifying angioid streaks necessitates a work-up for an underlying systemic disorder as well as close follow-up for potential ocular complications.
Idiopathic epiretinal membrane
Epiretinal membrane formation (macular pucker, premacular gliosis, premacular fibrosis, cellophane maculopathy) in young adults is often associated with an underlying etiology such as ocular trauma,76 posterior segment inflammation,77 combined hamartoma of the RPE and retina,78 Coats’ disease,79 and neurofibromatosis type II.80 Unlike in adults, idiopathic epiretinal membrane (Fig. 49.8) is rare in children and adolescents.81 Histopathologic studies demonstrate that epiretinal membranes consist of glial and retinal pigment epithelial cells, with a greater proportion of myofibroblasts and collagen formation observed in the epiretinal membranes of adolescents than in those of the elderly.82