Chapter 44 Abnormalities of Cone and Rod Function
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
Congenital color vision deficits as well as retinitis pigmentosa and its related disorders are covered in separate chapters (respectively, Chapter 10, Color vision and night vision, and Chapter 40, Retinitis pigmentosa and related disorders).
Disorders of the cone system
Achromatopsia
Congenital achromatopsia is a rare disorder, with an incidence of roughly 1 in 30 000.1 Individuals with this disorder have poor vision from birth and complain of poor color discrimination and photosensitivity, which relates to their visual acuity decreasing in bright light rather than actual discomfort. Patients with complete achromatopsia, also known as rod monochromatism, are generally considered to lack cones and have vision worse than 20/200. On the other hand, patients with incomplete achromatopsia, also known as atypical achromatopsia, may have slightly better visual acuity in the range of 20/80–20/200. Complete achromats have no color vision whereas incomplete achromats may have some residual color vision. Interestingly, because their color vision loss is congenital, even complete achromats may be able to identify colors. They generally perceive colors as lighter or darker shades of gray and may have learned that red is a darker shade of gray and yellow is a lighter shade of gray and so forth. Consequently, specific color vision tests (discussed below and in Chapter 10,) are designed to uncover this characteristic. Pendular nystagmus may be present, which can improve with age. Family history can be useful as achromatopsia displays an autosomal recessive inheritance pattern.
Diagnosis
On clinical examination, patients may have a normal fundus, or have subtle granularity or atrophy of the macula. The nerve may be normal or show some temporal pallor. Electrophysiologial testing is crucial to making the diagnosis in achromatopsia. Photopic and 30 Hz-flicker electroretinograms will show nearly or completely nonrecordable cone responses in the face of normal or near-normal rod responses (Fig. 44.1). Multifocal electroretinograms will be severely diminished. Full-field electroretinography (ERG) is critical to diagnosis as it helps differentiate achromatopsia from other entities that share a normal fundus exam and associated symptoms. For instance, an infant with Leber congenital amaurosis will also have nystagmus, extremely poor vision, and photosensitivity but will have extinguished photopic and scotopic ERGs. A blue cone monochromat will have poor vision, but will have recordable cone responses when stimulated with blue light on a yellow background. A patient with optic neuropathy would have optic nerve pallor with otherwise unremarkable fundus findings as well as decreased vision and color perception, but would show normal photopic and scotopic ERGs.
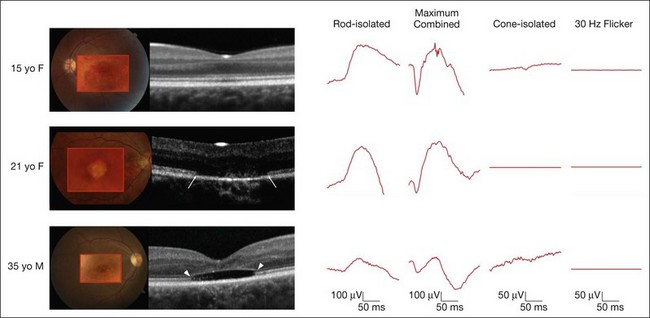
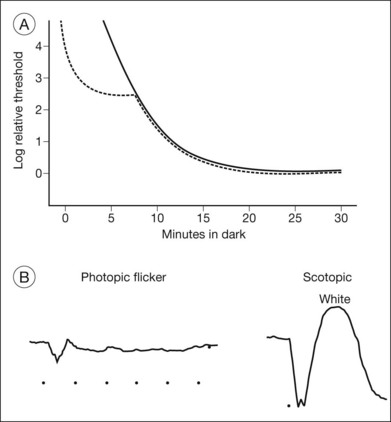
Other ancillary tests are also helpful in the diagnosis. Congenital achromats may be able to identify several of the pseudochromatic (Ishihara) color plates because they may have learned to distinguish colors as different shades of gray. Farnsworth D-15 testing may reveal a scotopic axis between the deutan and tritan axes. The Sloan achromatopsia test uses an achromat’s correlation of different shades of gray to various colors in order to distinguish them from normal individuals.2 Dark-adaptation curves will show a lack of a cone–rod break, reflecting the lack of cone function (Fig. 44.2). Visual field testing may reveal a small central scotoma but the peripheral visual fields should be either mildly constricted or normal, and, most importantly, they remain stable over time. Progression on serial visual field testing should raise suspicion for other disorders such as a cone–rod dystrophy or retinitis pigmentosa with a cone–rod pattern.
The ERG changes precede the less specific and sometimes more subtle findings seen on optical coherence tomography (OCT) (Fig. 44.1). A recent report calculated the frequency of these findings in a large series of 77 eyes of achromatopsia patients.3 Among the most obvious and common is disruption of the inner/outer-segment (IS–OS) junction occurring in 70% of these eyes. A hyporeflective, optically empty cavity may be seen in the cone layer of the foveola (60%), preferentially weighted to the nasal side of the foveal center, where there is higher density of cone photoreceptors. Also present in the majority of patients is foveal hypoplasia (83%). Disruption of the RPE and outer retinal atrophy can be seen in a minority of patients (18%). Although achromatopsia has traditionally been thought to be stationary, the imaging studies suggest that this view should be reconsidered.4 Progressive cone death may occur in achromatopsia patients with age as suggested by comparing the patient’s OCT to age-matched controls as well as a preferential age-dependence of features such as the hyporeflective zone, thinning of the outer nuclear layer, and RPE atrophy.3,5 Given the overlap in genetic and clinical findings, it is also plausible that some patients diagnosed with complete achromatopsia were incomplete achromats who later progressed.
Although the technique is not readily available in most clinics, adaptive optics scanning laser ophthalmoscopic imaging of the macular photoreceptor mosaic in a patient with complete achromatopsia has helped shed light on the pathophysiology.6 This case has revealed that the macular area has significant gaps within the photoreceptor mosaic, with a total absence of photoreceptors bearing the characteristics of normal healthy cones. These remaining photoreceptors appeared to have morphologic characteristics typical of rods.
Clinical molecular genetic testing for the four known genes responsible for the majority of achromatopsia cases is available from multiple laboratories (http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/clinical_disease_id/211350?db=genetests; accessed February 2012). A brief discussion of our molecular understanding of achromatopsia is available online.
Molecular basis of achromatopsia
Most cases of congenital achromatopsia are associated with four genes, which are primarily inherited in an autosomal recessive fashion. All mutations affect proteins involved in cone phototransduction. Two major mutations affect proteins that code for the alpha- and beta-subunit of cone cyclic-GMP gated (CNG) cation channels. CNGB3 codes for the beta-subunit and is responsible for approximately 45% of complete achromatopsia. It was originally identified in the Pingelapese people in the western Pacific nation of Micronesia.7 The second most-common mutation (25%) is in the CNGA3 gene, which codes for the alpha-subunit.8,9 The third gene that is associated with complete achromatopsia (<2%) is GNAT2, which codes for the alpha-subunit of cone transducin. Transducin is involved in the signal transduction cascade, and acts to close the cGMP channels.10,11 Finally, most recently, mutations in PDE6C have also been identified as a cause of achromatopsia (<2%).12 This gene, which has also been implicated in retinitis pigmentosa and congenital stationary night blindness, codes for the catalytic subunit of cone photoreceptor phosphodiesterase.
The vast majority of incomplete achromatopsia has been linked to missense mutations in CNGA3, which are inherited in an autosomal recessive fashion. At one time, it was felt that this was because mutations in CNGB3 and GNAT2 were primarily those leading to nonfunctional truncated products and thus only yielding the complete achromatopsia phenotype. However, the discovery of a frameshift mutation in GNAT1 that results in a truncated protein associated with an incomplete loss of color vision argues against this.13 As only a few of the mutations identified to date in CNGA3 are exclusive to incomplete achromatopsia while most mutations can also be associated with complete achromatopsia, additional epigenetic modifiers are being sought that may attenuate the color vision and visual acuity defects in incomplete compared to complete achromatopsia.
Treatment
There is currently no treatment for the underlying defects in achromatopsia. Photophobia can be reduced with tinted lenses, particularly orange or red lenses since rod photoreceptors are less sensitive to orange and red lights. Red-tinted soft contact lenses that transmit between 400 and 480 nm can reduce the stigma of having to wear dark glasses even indoors.14 Low vision aids may help to enhance visual acuity. Great strides have been made in research whereby adenoviral-mediated gene transfer has been used to correct the defect in all three genetic forms of human achromatopsia in corresponding animal models.15–17 As these gene-based therapies eventually move to human trials, identification of mutations in patients with achromatopsia will become crucial.
Cone monochromatism and blue cone monochromatism
Cone monochromatism is a congenital disorder in which two of the three cone systems (long- [L], middle- [M], and short-wavelength [S]) are absent or nearly absent. Monochromatism involving preservation of either the red or the green cone system is extremely rare, with an incidence estimated at 1 in 100 million.18 These patients have a normal visual acuity, no nystagmus and a normal ERG, residual red and green sensitive pigments by fundus densitometry, and spectral sensitivity curves similar to normal patients.19–21 Given its rarity, the mechanism remains poorly understood and is mentioned here mainly for completeness.
Diagnosis
Fundus examination may be normal, or may reveal progressive pigment irregularities and macular atrophy on ophthalmoscopy and autofluorescence.22–24 By history, symptoms, and visual acuity, these individuals resemble achromatopsia and thus there are several tests that may help the clinician distinguish between the two. Because they have functional S cones, these patients will have rudimentary dichromatic color discrimination and they can be distinguished from achromats based on the presence of residual tritan discrimination on color testing. One can also use Berson plates that have been specifically developed for this purpose.25–27 Although the photopic and 30 Hz cone flicker will be absent under normal testing conditions, an S-cone signal can be recorded with a blue light-flash on a yellow background.28 Scotopic and bright-flash ERGs can be normal or attenuated.22,28,29 The disorder has been reported to be either stable or progressive, and it is unclear whether this variability is due to the specific mutation, epigenetic influences, and/or environmental factors. Clinical molecular genetic testing is currently available for some of the mutations involved with blue cone monochromatism (see online for a discussion of molecular mechanisms).
Molecular basis of blue cone monochromatism
The genetic basis of blue cone monochromatism can be explained in light of the high degree of variability in the expression of L and M opsins. Humans have one L opsin gene and one or more M opsin genes, which sit in a tandem array on the X chromosome. The S opsin gene resides separately, on chromosome 7.30,31 To date, the primary mechanisms by which blue cone monochromatism occurs involve either a one-step or two-step mechanism.30,32,33 Approximately 40% of patients will have a one-step deletion that affects the locus control region, an upstream conserved sequence which regulates transcription such that only one opsin gene is expressed per cone photoreceptor.23,24,32 The other 60% have a two-step mutation process. The first step involves a nonhomologous recombination event between the L and M opsin genes, which reduces the number of genes in the opsin array on the X chromosome to one. A second mutation then occurs in the remaining opsin gene, rendering it ineffective.34 Not all cases are explained by mutations in the structure of the opsin array and it is likely that additional mechanisms will continue to be identified.32,33
Progressive cone dystrophies
These are inherited retinal dystrophies that usually present within the first three decades of life, although there are rare reports of patients presenting after the fifth decade.36–38 Decreased visual acuity and central scotomas, some degree of color vision loss, and photophobia are the usual symptoms. The visual defects become worse over time. Some are purely cone dystrophies in which rod function remains normal. In these individuals, night blindness is not a major complaint, nor is peripheral field loss. However, there are many patients who, sooner or later, will have diminished rod function and will manifest the accompanying symptoms at that time. Cone–rod dystrophies are primarily nonsyndromic, but can also be associated with syndromes including Bardet–Biedl and spinocerebellar ataxia, so attention must be paid to a review of systems and the medical history. All mechanisms of inheritance are possible with cone–rod dystrophies, and a careful collection of family history will assist with narrowing down the myriad of mutations responsible for this phenotype, as well as with genetic counseling.
Diagnosis
OCT may show thinning of the outer retinal layers, limited mostly to the central retina.39 Fundus autofluorescence can show central or parafoveal areas of increased or decreased autofluorescence with variable involvement of the periphery. However, the OCT and autofluorescence changes are nonspecific and may be similar in a number of other retinal degenerations as well. It is therefore important to remember that it is the electrophysiological evaluation coupled with these imaging studies which will help make the definitive diagnosis.40
Although not readily available in most clinics, adaptive optics imaging has been used to investigate eyes with cone dystrophy. With this technique the photoreceptor mosaic shows increased cone spacing in cone dystrophy compared to normal subjects or retinitis pigmentosa patients.41
The diversity of mutations responsible for cone and cone–rod dystrophies currently precludes widespread screening for mutations. However, this may change as more efficient screening mechanisms such as microarrays become more readily available.42 A brief discussion of the molecular mechanisms of cone–rod dystrophies is given online.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


