13 The arterial circulation of the retina, with the exception of the cilioretinal artery, is supplied by the central retinal artery. The quadrantal branches divide upon leaving the optic disc and extend toward the periphery; at the ora serrata, they turn back toward the optic disc to form venules. In 1977, Awan conducted a study on the incidence of nonaneurysmal and noncommunicating anomalies of the retinal arteries in 2100 normally functioning eyes of healthy subjects. Unusual anomalies included triple branching, anomalous course, arteriolar–arterial crossing, unusual tortuosity, prepapillary loops, aberrant macular arteries, unusual supply of the optic disc, presumed total ciliary arterial supply of the retina, anomalous relationship with the central retinal vein at the optic disc, and pseudoaneurysm of a major retinal artery (Fig. 13–1).1 In one study, the incidence of cilioretinal artery was found to be as high as 49.5%. It is considered the most common congenital vascular anomaly of the retina.2 Retinal telangiectasis is a retinal vascular anomaly characterized by irregular dilation and incompetence of the retinal vessels.3 These vascular anomalies may be confined to the juxtafoveal area, a condition termed idiopathic juxtafoveal telangiectasis. Central vision loss may result because of exudation or diffusion abnormalities from ectatic and incompetent juxtafoveal retinal capillaries. The condition may be congenital or of unknown cause and may be bilateral or unilateral. Idiopathic juxtafoveal retinal telangiectasis is classified into several groups.3 In 1982, Gass and Blodi4 proposed a classification system of idiopathic juxtafoveolar retinal telangiectasis based on biomicroscopic and fluorescein angiographic findings. There were subsequent revisions in 1987 and 1992 (Table 13–1). Patients are divided into three groups and are further subdivided into group 1A and 1B. Group 1A has visible and exudative idiopathic retinal telangiectasis. Most of these patients are males with unilateral disease. The temporal macula is typically involved with exudation present. The size of the involved area is two disc diameters (DD) or greater. This is considered a mild form of Coats disease that may progress to the entire retina and cause massive exudation. Telangiectasis involves the superficial and deep capillary plexus of juxtafoveolar retinal capillaries. Vision loss occurs secondary to the extension of exudation into the foveolar area. Laser treatment to these telangiectatic capillaries is effective in reducing the foveolar exudation and in improving or preserving visual acuity. No systemic disease is associated with this category. Group 1B is identical to group 1A in patient characteristics. The difference between group 1A and 1B is in the size of the affected area, less than 2DD. Visual acuity is rarely affected. Group 2 is subdivided into group 2A and 2B. The telangiectasis is acquired during middle age, has no sex predilection, and presents bilaterally in 98% of cases. Group 2A has occult and nonexudative idiopathic juxtafoveolar retinal telangiectasis, the most common form of the disease. In many patients, slight loss of retinal transparency is the only biomicroscopic clue to the presence of these capillaries. Fluorescein angiography is the best method to detect the telangiectatic vessels. The temporal parafoveolar area is involved in all cases, within 1 DD of the center of the fovea. Minimal loss of vision is secondary to foveolar atrophy and rarely is due to subretinal neovascularization. Forty-five percent of these patients have golden refractile deposits near the inner retina surface in the juxtafoveolar area. The cause of these deposits is unknown. The findings in group 2A eyes are further subdivided into five stages of development (Table 13–2). Group 2B patients have a juvenile onset of occult familial idiopathic juxtafoveolar retinal telangiectasia. Gass and Blodi included the case of two brothers with bilateral subretinal neovascularization and subtle retinal juxtafoveolar telangiectasis in this category. These patients differ from group 2A patients in that they lack right-angle venules, superficial retinal refractile deposits, and stellate pigmented plaques.
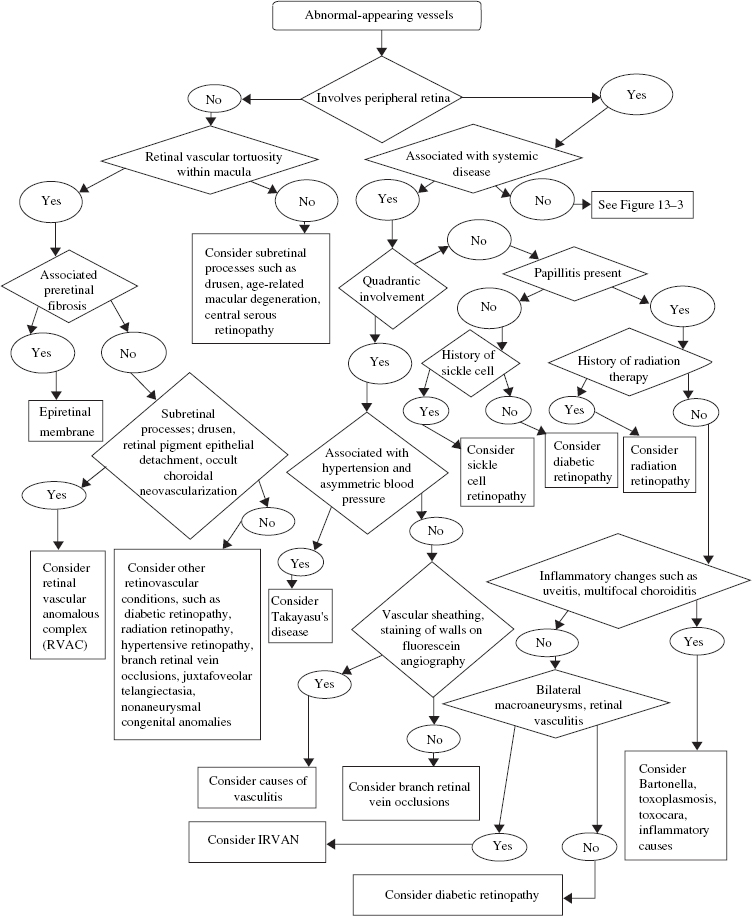
Abnormal-Appearing Retinal Vessels
Congenital Tortuosity of the Retinal Vessels (Nonaneurysmal Congenital Anomalies)
What Are Arterial Vascular Anomalies?
WHAT IS THE MOST COMMON CONGENITAL VASCULAR ANOMALY OF THE RETINA?
What Is Idiopathic Juxtafoveal Retinal Telangiectasis?
WHAT IS THE CLASSIFICATION SYSTEM FOR IDIOPATHIC JUXTAFOVEOLAR RETINAL TELANGIECTASIS?
WHAT ARE THE CHARACTERISTICS OF GROUP 1 OF GASS AND BLODI’S CLASSIFICATION?
What Are the Characteristics of Group 2 of Gass and Blodi’s Classification?

| Stage | Biomicroscopy | Fluorescein Angiography |
|---|---|---|
| Stage 1 | No abnormality | Early: no capillary dilation Late: mild staining at the outer temporal parafoveolar area |
| Stage 2 | Perifoveolar graying Minimal or no telangiectasia | Early: telangiectasia of the outer temporal capillary network |
| Stage 3 | Dilated/blunted retinal venules extending into parafoveolar retina | Capillary dilation and permeability changes |
| Stage 4 | Foci of hyperplastic RPE beneath Right-angle venules leading to an irregular or stellate plaque | Capillary dilation and permeability changes |
| Stage 5 | Subretinal neovascularization in the temporal parafoveolar area associated with intraretinal pigment epithelial migration | |
RPE, retinal pigment epithelium.
WHAT ARE THE CHARACTERISTICS OF GROUP 3 IN GASS AND BLODI’S CLASSIFICATION?
Group 3 is subdivided into 3A and 3B. Patients in group 3A have extensive capillary occlusion associated with juxtafoveolar telangiectasis. Given the large capillary-free zones (greater than 1 DD in size), the reason for continued good visual acuity is unclear. Systemic disease (hypertension, diabetes, or cardiovascular disease) is present in all described cases.
Group 3B consists of patients with occlusive idiopathic juxtafoveolar retinal telangiectasis associated with central nervous system vasculopathy. All have loss of central vision associated with the telangiectasis, capillary closure, and minimal exudation. A specific central nervous system lesion was never identified. Gass and Blodi suspected that these patients may have had an autosomally dominant cerebroretinal vasculopathy.4–7 Overall, visual loss in Group 3 is presumed to be caused by ischemia from the capillary occlusion rather than from exudation.
HOW IS IDIOPATHIC JUXTAFOVEOLAR TELANGIECTASIA TREATED?
In patients in group 1, this condition is considered nonfamilial and predominantly exudative. Laser photocoagulation is recommended to stabilize and prevent further visual loss. In groups 2 and 3, the condition is nonexudative, obstructive, and occasionally familial, and patients benefit little from laser photocoagulation treatment. Most patients have a good longterm prognosis for retaining reading vision in at least one eye.
What Are Retinal Vascular Anomalous Complexes (RVAC)?
Retinal vascular anomalous complexes (RVACs) are angiomatous lesions within the deep retina that are derived from the retinal circulation and associated with age-related macular degeneration (AMD).8 When first described, they were identified with pigment epithelial detachments,9 but subsequent studies have shown this to be uncertain.10,11 Over time, they may become chorioretinal anastomoses.
Takayasu Disease
What Is Takayasu Disease?
Takayasu disease, or occlusive thromboaortopathy or pulseless disease, is an idiopathic, inflammatory disease of the large arteries, including the aorta and its branches, that leads to arterial narrowing and obliteration. The four major complications include Takayasu retinopathy, secondary hypertension, aortic regurgitation, and aortic or arterial aneurysm.12 Studies of retinal hemodynamics suggest that carotid artery involvement leading to diminished ocular perfusion is the mechanism of Takayasu retinopathy.
In Japan, women account for 90% of the cases of Takayasu arteritis. The female-to-male ratio decreases, however, as one moves toward the West.13 Genetic studies have revealed a strong human leukocyte antigen (HLA) association with the HLA A24-B52-DR2 haplotype. Patients with this haplotype are thought to be more likely to develop accelerated inflammatory progression and resistance to steroid therapy.14
What Are the Signs and Symptoms of Takayasu Disease?
The clinical manifestations will vary depending on the site and severity of the affected vessels. The aorta and its tributaries, including the braciocephalic, carotid, subclavian, vertebral, renal, coronary, and pulmonary arteries, are narrowed.
Patients may present with complaints of dizziness, syncope with or without headache, and visual disturbances. More commonly, weak or absent radial pulses are demonstrated. A comparison of the blood pressure in each arm is classically asymmetric. On examination, patients may have an associated systolic vascular murmur (aortic regurgitation secondary to dilatation of aortic valve root); this may lead to congestive heart failure and arrhythmia. Patients at risk of developing Takayasu retinopathy will have a prolonged arm-to-retina circulation time.
On laboratory evaluation, patients may have elevated acute-phase reactant markers, including erythrocyte sedimentation rate (ESR) and elevated C-reactive protein.
What Is the Pathogenic Mechanism Involved in Takayasu Disease?
Takayasu disease is a chronic vasculitis of unknown cause. On pathologic examination, Takayasu disease is a panarteritis involving the intima, media, and adventitia; this finding produces an array of ischemic clinical symptoms attributable to stenosis or secondary thrombus formation within the vasculature. Its various clinical manifestations are due mainly to insufficient perfusion of the tissues caused by progressive narrowing and obliteration of their vascular supply.15
The retinal vascular changes in Takayasu disease occur in response to a gradual decrease in the central retinal artery pressure below a critical level. The retinal vessels react to diminished perfusion by generalized vasodilation, microaneurysm formation, and hyperpermeability of the vessel wall. A further decrease in arterial pressure peripheral to the shunt vessels accelerates vascular nonperfusion, leading to vessel occlusion with secondary neovascularization of the iris, chamber angle, and retina.
What Are the Ocular Findings Associated with Takayasu Arteritis?
Retinal vascular changes occur secondary to chronic decrease in the central retinal artery pressure. These retinal changes include progressive small vessel dilation, microaneurysm formation, retinal arteriovenous anastomoses, vitreous hemorrhage, proliferative retinopathy, vitreous hemorrhage, anterior ischemic neuropathy, and retinal emboli.16–18 Retinal arteriovenous (AV) shunt formation is a unique retinal finding in moderate to advanced stages of Takayasu disease19 and may be followed by capillary nonperfusion in the midperipheral retina.
There are three reported types of retinal AV shunts. One type forms at AV crossings; the other develops consequent to capillary dilatation followed by preferential channel formation. In the more advanced cases of Takayasu disease, peripapillary vascular loops have been reported; however, the midperipheral retina remains the major site of AV shunt formation.20
Patients also sometimes demonstrate dilated episcleral vessels with intravascular sludging and signs of anterior and posterior uveitis with anterior chamber cells. Dilation is noted in the retinal veins with impressive intravascular sludging of blood. This is more prominent on fluorescein angiography, which highlights numerous capillary microaneurysms seen as dot hemorrhages on indirect ophthalmoscopy.
How Is Takayasu Disease Diagnosed?
Imaging studies, including magnetic resonance imaging (MRI), computed axial tomography (CT), or digital subtraction angiography, may demonstrate narrowing of the aorta and its tributaries. Decreased central retinal artery pressure with increased arm-to-retina circulation time confirms the diagnosis.
On examination, patients may have absent or faint radial pulses and asymmetric blood pressure with possible systolic murmur on cardiac auscultation.
What Is the Visual and Systemic Prognosis of Takayasu Disease?
If diagnosed and treated at an early stage, before ischemic symptoms develop, the prognosis is positive. The high incidence of cerebrovascular accidents due to renovascular hypertension has been reported as the main cause of death in some Asian countries, including China, Thailand, and India.
Takayasu retinopathy regresses with resolution of carotid artery stenosis, leading to a gradual increase in the central retinal artery pressure.
How Is Takayasu Disease Treated?
Initially, Takayasu arteritis is managed with high-dose steroids, followed by maintenance with low-dose steroids and aspirin. In cases of steroid resistance, immunosuppressive drugs are used as supplementary treatment.
In cases of Takayasu retinopathy, once the retinal artery pressure rises above a critical level, the AV shunts and other retinal findings slowly regress. Early diagnosis and treatment prior to ischemic manifestations is primary in management of the disease.
Eales Disease
What Is Eales Disease?
Eales disease is associated with idiopathic obliterative peripheral retinal vasculopathy and retinal periphlebitis. It is uncommon in North America but is a major cause of widespread visual loss on the Indian subcontinent. Healthy young adult males in their second to third decade of life are primarily affected. In most patients, ocular findings are bilateral. Although there is no evidence of a hereditary tendency, studies have shown a statistically significant higher phenotype frequency of HLA B5 (B51), DR1, and DR4 among patients with Eales disease compared with controls.21
What Is the Pathogenesis of Eales Disease?
Eales disease is also known as periphlebitis retinae; evidence suggests that the inflammatory aspect of this disease affects the arterioles as well as the venules.22 Sheathing of peripheral retinal vessels occurs secondary to the leukocytic infiltration of the vessel wall, with a resultant perivascular cuff of lymphocytes and plasma cells. Early capillary closure of the temporal peripheral retina is followed by retinal ischemia, leading to neovascularization between areas of the perfused and nonperfused retina. In most patients, there is a strong fibroglial component to this neovascular tissue that may lead to recurrent vitreous hemorrhages and tractional retinal detachment.
What Are the Retinal Manifestations of Eales Disease?
In the early stages of Eales disease, patients develop ocular inflammation, manifested by vascular sheathing of varying degrees. On fluorescein angiography, areas of sheathing demonstrate hyperfluorescence; however, the intensity of this hyperfluorescence does not correlate with the intensity of inflammation. Ocular inflammation may also present as keratic precipitates, anterior chamber cell and flare, vitreous cells, and cystoid macular edema. Intraretinal hemorrhages with microvascular occlusion lead to vascular tortuosity and retinal ischemia, mainly in the temporal retina. Fluorescein angiography highlights venous beading, microaneurysms, and AV shunts at the junction of the perfused and nonperfused retina.
What Are the Systemic Manifestations of Eales Disease?
Eales disease can affect the vestibuloauditory system, with resultant sensorineural hearing loss in up to 50% of patients.23 In several reports, Eales disease has been associated with cerebrovascular accidents.24
How Is Eales Disease Diagnosed?
Although Eales disease is distinct, its diagnosis remains one of exclusion. Patients should be evaluated for other systemic diseases leading to retinal vascular abnormalities, such as sickle cell disease, diabetes, sarcoidosis, and connective tissue disorders such as systemic lupus erythematosus. The presence of active pulmonary tuberculosis should be excluded, and the tuberculin hypersensitivity status should be determined. Patients should be questioned extensively with regard to hearing and balance problems. According to a recent study, Eales disease may be associated with factor V Leiden mutation and previous thrombotic events. This finding has caused the standard evaluation of Eales disease and other causes of peripheral retinal neovascularization to include factor V Leiden testing and further coagulopathy evaluation.25
Branch retinal vein occlusion (BRVO) is commonly confused with Eales disease. The distinguishing feature in Eales is the involvement of multiple retinal quadrants without respect to the retinal midline.
What Is the Relationship between Eales Disease and Tuberculosis?
Many studies have shown a higher than normal incidence of tuberculin hypersensitivity in patients with Eales disease.26,27 In one study, this incidence was as high as 48%.28 Tuberculin hypersensitivity may play a role in the pathogenesis.
What Is the Visual Prognosis for Eales Disease?
Eales disease affects mainly the peripheral retina while sparing the macular vasculature. Therefore, many patients retain good central visual acuity that is interrupted by periods of blurred vision and floaters caused by vitreous hemorrhage. Vitreous hemorrhage remains the most common cause of visual loss. Complications such as persistent vitreous hemorrhage, traction retinal detachment, neovascular glaucoma, cystoid macular edema, macular holes, retinal telangiectasia, or epiretinal membrane formation may cause long-term visual loss.
How Is Eales Disease Treated?
Retinal neovascularization is very responsive to peripheral laser photocoagulation. In patients with persistent vitreous hemorrhage, pars plana vitrectomy is recommended and yields good results.27,28 Retinal detachments in Eales disease are treated with standard scleral buckle and vitrectomy techniques. If possible, preoperative laser to areas of attached peripheral avascular retina should be performed prior to vitrectomy. There are no treatment modalities to prevent or reverse the initiating retinal ischemia.
Idiopathic Retinal Vasculitis, Aneurysms, and Neuroretinitis (IRVAN) Syndrome
What is IRVAN Syndrome?
Originally described by Kincaid and Schatz,29,30 this syndrome is characterized by the presence of bilateral retinal vasculitis, multiple macroaneurysms, neuroretinitis, and peripheral capillary nonperfusion. More recently, Chang and colleagues31 proposed the acronym IRVAN to highlight the most prominent clinical features of this syndrome (idiopathic retinal vasculitis, aneurysms, and neuroretinitis). It typically affects young, healthy persons and appears to have a female predilection. Family history and extensive systemic investigations are negative. Patients are frequently asymptomatic at the time of presentation despite the impressive fundus appearance on clinical examination.
What Are the Retinal Findings in IRVAN?
Reported cases show focal areas of vascular sheathing. Numerous aneurysmal dilatations of the retinal arterioles are noted at the optic nerve head, on the macular branch arteries, and at branching sites of the first- and second-order retinal arteries. These vascular abnormalities typically have a “Y-shaped” configuration and also have been described as “knots” tied onto the arteriolar tree.32–34
Patients may have bilateral optic nerve head edema with surrounding peripapillary exudates extending to the macula. Areas of peripheral capillary nonperfusion may be associated with retinal neovascularization and vitreous hemorrhage (Table 13–3).
What Are the Fluorescein Angiographic Findings in IRVAN?
Fluorescein angiography reveals prominent vascular dilatation with evidence of late staining of macroaneurysms and retinal arteriole walls. The peripheral retina demonstrates extensive areas of capillary nonperfusion. The optic nerve head commonly appears hyperfluorescent with extensive leakage in the late stages of the angiogram.
What Is the Visual Prognosis for IRVAN?
Exudative retinopathy and peripheral capillary nonperfusion may threaten vision. Exudative retinopathy is typically concentrated in a peripapillary area adjacent to the retinal and optic nerve head aneurysms.
| Anterior segment | Mild anterior uveitis |
| Anterior vitreous | Cellular debris |
| Lens | Posterior subcapsular cataract |
| Retina | Tortuous, telangiectatic retinal arteries, aneurysms |
| Retinal edema and circinate exudation | |
| Serous retinal detachment | |
| Tractional band detachment | |
| Macula | Macular exudates |
| Peripheral retina | Marked vessel attenuation |
| Extensive retinal ischemia | |
| Optic nerve | Optic nerve head edema |
| Optic nerve head aneurysms “hairpin loops” | |
| Tufts of neovascularization | |
| Vitreous | Recurrent vitreous hemorrhages |
IRVAN, idiopathic retinal vasculitis, aneurysms, and neuroretinitis.
Extensive retinal ischemia secondary to peripheral capillary nonperfusion often leads to retinal neovascularization with subsequent vitreous hemorrhage. In rare cases, ischemia leads to iris neovascularization with neovascular glaucoma.
How Is IRVAN Treated?
Despite the presumed inflammatory component of this syndrome, patients do not benefit from oral or intravenous high-dose steroids. Given the adverse side effects of systemic steroids, this therapy is not recommended. One case report noted the disappearance of retinal aneurysms over a 7-year interval without surgical intervention.33
The role of panretinal photocoagulation in treating other ischemic retinopathies may be applied to IRVAN. Although the timing of laser photocoagulation in treating the associated retinal ischemia is not well defined, it is beneficial in stabilizing anterior segment and retinal neovascularization.
In cases with recurrent vitreous hemorrhage, pars plana vitrectomy with supplemental endolaser may be required to stabilize the disease. At present, proposed treatments for this syndrome are recommended for symptomatic patients.
How Is IRVAN Different from Eales Disease?
Although both IRVAN and Eales disease are characterized by vasculitis, Eales tends to affect venules more commonly than arterioles. Eales also occurs in young healthy males with associated tuberculin hypersensitivity, vestibuloauditory deficits, and rare cerebral infarction.
Sickle Cell Disease
What Is Sickle Cell Disease?
Heterozygous and homozygous forms of sickle cell disease involve mutations in the hemoglobin gene resulting in the production of defective or abnormal peptide chains of the hemoglobin molecule.
Normal hemoglobin, HbA, has two alpha and two beta chains. In HbS, or sickle hemoglobin, valine replaces glutamic acid on the beta chain. HbC has lysine substituting for glutamic acid on the beta chain. HbSS is the homozygous form of sickle cell disease; the heterozygous states are HbAS, HbSC, and alpha or beta thalassemia with failed or defective production of one peptide chain in the hemoglobin molecule.
About 8% of black Americans are heterozygous for HbS. The gene frequency is highest in central Africa, particularly in areas where malaria is endemic. Heterozygotes gain mild protection against falciparum malaria.
What Is the Pathogenic Mechanism Involved in Sickle Cell Disease?
Genetic mutation produces hemoglobin molecules that polymerize under acidic or hypoxic conditions, causing the formation of sickled erythrocytes. These sickled red blood cells have rigid membranes, leading to difficulties in the microcirculation. Vasoocclusions in all organ systems are possible.
The erythrocyte polymerization rate depends on the intracellular concentration of HbS and the extent of deoxygenation. Factors such as acidosis or increased red blood cell 2,3-diphosphoglycerate lower the oxygen affinity of red blood cells and enhance polymerization and sickling.
What Are the Signs and Symptoms of Sickle Cell Disease?
Patients with HbSC and thalassemia demonstrate the most severe ocular manifestations, whereas HbSS patients exhibit the most dramatic systemic complications. Heterozygotes for sickle cell require a much lower oxygen tension for sickling than homozygotes. Overall, heterozygotes have a higher life expectancy and lower morbidity compared with sickle cell disease patients.
The constitutional manifestations of sickle cell anemia include failure to thrive and susceptibility to infections (especially due to encapsulated organisms). The vasoocclusive manifestations caused by microinfarcts affect various parts of the body, particularly the abdomen, chest, back, and joints.
In homozygote SS patients, anemia becomes severe if erythropoiesis is suppressed. Aplastic crises in these patients may be caused by folic acid deficiency and infection (especially with parvovirus).
Nonocular signs and symptoms, including musculoskeletal and joint pain, abdominal discomfort, cholelithiasis, and episodic anemia seen commonly in HbSS patients are rare in those with HbSC disease.
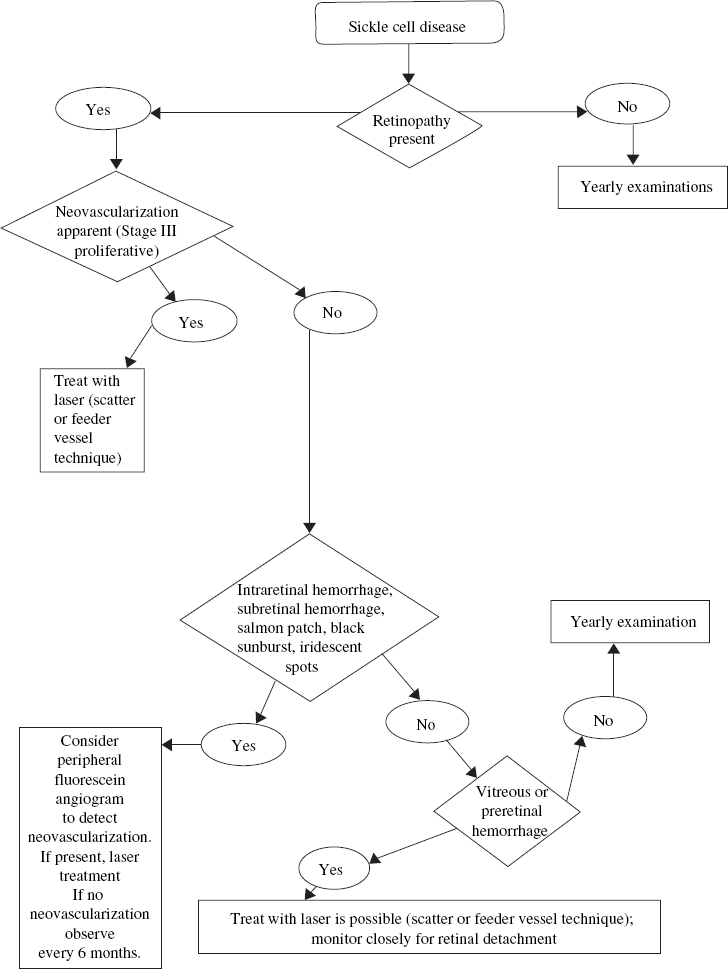
What Are the Ocular Complications of Sickle Cell Disease?
The comma sign of the conjunctiva refers to changes in the conjunctival vessels’ appearance that occur secondary to microvascular occlusions. The disc sign of sickling refers to intravascular occlusions of the small surface vessels of the optic nerve that do not result in visual impairment and do not appear to impede perfusion substantially. Neovascularization of the disc is rare.
Patients also demonstrate an enlarged foveal avascular zone, macular vascular occlusion, branch retinal artery occlusion (BRAO), and, rarely, spontaneous central retinal artery occlusion (CRAO). These manifestations may lead to central, paracentral, or even complete loss of vision34,35 (Table 13–4).
Nonproliferative and proliferative retinal changes occur in sickle cell disease. A nonproliferative change is the occurrence of a salmon-patch hemorrhage, a focal intraretinal and subretinal hemorrhage that develops after an acute arteriolar occlusion, typically in the midperipheral retina, with subsequent ischemic necrosis and hemorrhage through the vessel wall. The term salmon patch comes from the orange discoloration that develops in this area after several days. Over time, the hemorrhage is resorbed from the intraretinal space and a schisis cavity remains. Iridescent spots within the schisis cavity are thought to represent collections of macrophages responsible for resorption and breakdown of the preretinal hemorrhage.
| Conjunctiva | Comma sign |
| Optic nerve | Intravascular occlusions of small surface vessels of the optic nerve (“disc sign” of sickling) |
| Macula | Enlarged foveal avascular zone (FAZ) Macular arteriolar occlusions (infrequent) Epiretinal membranes Macular holes |
| Retina | Venous tortuosity “Salmon-patch” hemorrhage Iridescent spots Schisis cavity “Black sunburst” Angioid streaks Central retinal artery occlusions Branch retinal artery occlusions Neovascularization in the periphery Arteriovenous shunts in periphery |
| Choroid | Choroidal vascular occlusion |
Black sunbursts are black chorioretinal scars that represent focal areas of hyperplastic retinal pigment epithelium (RPE) in areas of previous ischemia and hemorrhage. These scars are characteristically perivascular in location and do not produce clinical symptoms. They are sometimes mistaken for focal areas of chorioretinitis.
Proliferative sickle cell retinopathy primarily tends to affect the temporal aspect of the peripheral retina. It may be more progressive in children and adolescents than in adults.
Angioid streaks occur more commonly in patients with HbSS disease compared to patients with HbSC disease. Rarely, angioid streaks may be associated with choroidal neovascularization occurring as a result of trauma.
Choroidal vascular occlusions may result from posterior ciliary artery occlusions and appear as triangular, hypopigmented patches at the level of the RPE. These are not thought to be visually significant.
How Is Sickle Cell Anemia Diagnosed?
The diagnosis of sickle cell anemia should be considered in patients with African heritage and hemolytic anemia. Similarly, any patient with African heritage who presents to the ophthalmologist with a hyphema should be tested for sickle cell disease.
The peripheral blood smear reveals normochromic normocytic red blood cells with some target cells. The presence of Howell–Jolly bodies, siderocytes, and occasional normoblasts suggests defective splenic function.
The diagnosis of the sickle cell trait or any sickle cell syndrome depends on demonstration of sickling under lowered oxygen tension in the sickle prep test. If the sickle prep is positive, hemoglobin electrophoresis is performed to distinguish between the homozygous and heterozygous forms of the disease.
How Is Sickle Cell Disease Treated?
Before birth, fetal hemoglobin or HbF, is the predominant form of hemoglobin. At birth, the switch from fetal to adult hemoglobin is not complete such that normal adults have about 1% HbF. It has been proven that HbF interferes with HbS polymerization.36
Understanding this pathophysiology has led investigators to treat sickle cell disease by increasing the distribution and concentration of fetal hemoglobin in sickle erythrocytes. Drugs such as hydroxyurea (HU) have laboratory and clinical efficacy in HbSS disease, and a recent study demonstrated its therapeutic benefits in pediatric patients with severe HbSC disease.37 Larger clinical trials of HU therapy in HbSC disease are warranted.
What Is the Pathogenesis of Sickle Cell Retinopathy?
Early forms of angiogenesis are seen in 40% of HbSS disease compared with 80% of HbSC disease.38 Sickle cell retinopathy is a disease that affects primarily the peripheral retinal vasculature. Peripheral vascular occlusion secondary to sickled erythrocytes results in a nonperfused and ischemic peripheral retina.39 Neovascularization develops at the junction of the perfused and nonperfused retina.
Vasoocclusions and subsequent vascular remodeling causes formation of AV anastomoses. It is at these anastomotic channels that preretinal neovascular formations, called sea fans, form. Several studies have also reported formation of sea fans at hairpin loops and AV crossings.40 The most common sites of sea-fan formation are in the superotemporal and inferotemporal peripheral retina. With time, most sea fans autoinfarct and regress; rarely, they lead to vitreous hemorrhages, and produce traction retinal detachments. Sea fans are the predominant causes of blindness in sickle cell disease.
Occlusive disease of the perifoveal arterioles is known to occur in sickle cell disease.41 An enlargement of the foveal avascular zone and ischemic maculopathy may result.
What Is the Classification System for Proliferative Sickle Cell Retinopathy?
Proliferative sickle retinopathy is most prevalent in patients with HbSC and S-thalassemia disease. In general, sickle cell trait and HbAS do not cause systemic disease or retinal neovascularization.
In 1971, Goldberg proposed a classification system for the development of proliferative sickle retinopathy (Table 13–5). Stage I refers to peripheral arteriolar occlusions seen as “silver-wire” vessels with an avascular zone of retina anterior to the occluded vessels on fluorescein angiography. Stage II is the vascular remodeling that takes place with the formation of AV anastomoses at the junction of vascular and avascular retina. On fluorescein angiography, these AV anastomotic channels do not leak.
| Stage I | Peripheral arteriolar occlusion |
| Stage II | Peripheral arteriolar–venular anastomoses |
| Stage III | Neovascular proliferation |
| Stage IV | Vitreous hemorrhage |
| Stage V | Retinal detachment |
Stage III represents the appearance of neovascularization from the AV anastomotic channels at the border of the perfused and nonperfused retina. These neovascular tufts demonstrate dye leakage on fluorescein angiography and have a characteristic fanshaped appearance. With time, these sea fans may acquire surrounding fibroglial tissue.
Stage IV refers to the vitreous hemorrhage that occurs from peripheral neovascular fronds secondary to minor ocular trauma or vitreous contraction. Previous studies found that the associated risk factors for vitreous hemorrhage are HbSC disease, more than 60 degrees of perfused sea fans, and the presence of old hemorrhage in the vitreous that can lead to vitreous band formation, secondary vitreous traction, and subsequent vitreous hemorrhage.
Stage V represents tractional retinoschisis or tractional retinal detachment secondary to vitreoretinal traction bands and membranes. These bands may cause rhegmatogenous retinal detachments from full-thickness retinal break formation.
More commonly, the neovascular tufts tend to involute and regress with time and rarely lead to severe visual impairment.42 Autoinfarction is thought to occur when the feeder vessel occludes. The mechanism for involution is possibly due to ischemia from fibroglial tissue strangulation of the feeder vessels.43
What Are the Local and Systemic Angiogenic Factors Associated with Proliferative Sickle Cell Retinopathy?
A study by Cao and colleagues demonstrated that autocrine production of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) are associated with sea-fan formation.44 This finding supported the previous hypothesis that local rather than systemic angiogenic factors are responsible for neovascular changes in sickle cell patients.
How Is Proliferative Sickle Cell Retinopathy Treated?
The more severe stages of proliferative sickle cell retinopathy are more common in children and adolescents, especially those with HbSC and S-thalassemia. Hence, the goal of treatment is early detection of neovascularization and eradication of new vessels before progression to more advanced stages of proliferative sickle cell retinopathy.
Treatment is not recommended for stage I and stage II disease. In the past, various treatment options, including diathermy, cryotherapy, xenon-arc photocoagulation, and argon-laser photocoagulation have been used to treat peripheral retinal neovascularization. Today, laser photocoagulation to the area of peripheral neovascularization is most commonly used. Techniques vary widely and include feeder vessel photocoagulation, direct treatment of the neovascular frond, and treatment of the nonperfused ischemic retina anterior to neovascularization (Fig. 13–2). Ease of administration and fewer complications following scatter laser photocoagulation led Jampol and coworkers to recommend this approach as the initial therapy in most cases.45–47
Retinal occlusive events may be treated by exchange erythrocyte transfusion in patients with bilateral retinal vascular occlusion.48 A follow-up case report in a 9-year-old boy with extensive perimacular arteriolar occlusions, however, did not demonstrate visual improvement, possibly secondary to treatment delay.49 Therefore, further studies regarding the role of exchange erythrocyte transfusion and visual outcomes in the setting of macular and retinal occlusive change are warranted.
Wyburn–Mason Syndrome
Arteriovenous Malformations of the Retina
WHAT ARE ARTERIOVENOUS MALFORMATIONS OF THE RETINA?
Arteriovenous malformations (AVMs) result from the persistence of an embryonic vascular pattern in which arteries or arterioles communicate directly with veins or venules. Numerous terms have been used to describe this lesion, including racemose aneurysm, racemose angioma, AV aneurysm of the retina, retinal arteriovenous fistula, racemose hemangioma, cirsoid aneurysm, aneurysma racemosum retinae, congenital AV anastomosis, and aneurysmal varix.
The vascular malformation may range in size from one AV communication to a complex anastomotic system with indistinct arterial and venular components. There is a predilection for the papillomacular bundle and the temporal quadrants of the retina. AVMs differ from von Hippel-Lindau disease, which has an autosomal-dominant pattern.
WHAT ARE THE OCULAR COMPLICATIONS ASSOCIATED WITH RETINAL ARTERIOVENOUS MALFORMATIONS?
Arteriovenous malformations may cause macular, preretinal, and vitreous hemorrhages, as well as central retinal vein occlusion (CRVO) and BRVO. Venous occlusive disease is thought to be associated with the spontaneous regression or remodeling of the AVM. Rare cases of secondary neovascular glaucoma have also been reported.50,51
What Is the Classification System of the Various Arteriovenous Malformations in the Retina?
In 1973, Archer and associates classified retinal AVMs into three groups.52 Group 1 is characterized by an intervening abnormal capillary bed between the communicating artery and vein. The AVM is localized to one sector of the retina, often the macular area, and is usually an incidental finding on ophthalmoscopic examination. These anomalous communications rarely decompensate, and there is no fluorescein leakage; rarely, they involve the peripapillary structures. Association with other ocular anomalies or significant cerebrovascular malformations is uncommon, and visual acuity is not normally affected. There is no indication for treatment, and further evaluation for cerebral AVMs should not be pursued.
Group 2 describes AVMs with direct communication and without intervening capillary plexus or arteriolar elements. These may be single end-to-end junctions or multiple anastomoses with channels that are intermediate (100 to 200 μ) or large (200 to 300 μ) size. The afferent vessels may demonstrate beading and multiple fusiform dilatations within their walls with occasional aneurysm formation. Surrounding capillary beds may show dropout or nonperfusion.
These anomalous communications commonly remain stable over time, with rare instances of regression, thrombosis, and remodeling. Decompensation of the AVM may demonstrate fluorescein leakage, retinal edema, intraretinal exudates, and hemorrhage. In some instances, photocoagulation may be used to decrease the caliber of the afferent artery. Association with cerebral AVMs is uncommon. Carotid or cerebral angiography is not advisable unless adequate clinical suspicion exists.
Group 3 refers to AV communications that are extensive, complex, and have secondary retinal complications leading to severe visual impairment at an early age. The arterial and venous components of these AVMs are difficult to separate. The anastomosing channels are large in caliber (500- to 600-μ diameter) and cover most of the fundus; they develop fibromuscular coats within the media of the vessel, with a surrounding adventitial covering. The intervening retina may be attenuated with cystic degeneration.
Various degrees of vascular decompensation occur. Cerebral AVMs, the most common of these malformations, are part of this group.52