9
Posterior Uveitis and Panuveitis
John J. Huang and Paul A. Gaudio
INTRODUCTION
Posterior uveitis encompasses inflammation of the choroid, retina, vitreous, and retinal vasculature. By definition, posterior uveitis involves some morphological change visible on examination of the retina and choroid, usually in the form of one or many spots or patches. “Spillover” inflammation into the anterior segment with associated anterior chamber cells can often be seen, and when the degree of anterior chamber inflammation is significant, the process is termed diffuse uveitis or panuveitis—these two terms are synonymous. Posterior uveitis has a high association with systemic infections and autoimmune diseases.
By far, the most common infectious posterior uveitis is due to toxoplasmosis, while herpetic retinitis is a distant second (Table 9.1). Immune-mediated posterior uveitides are many, and occur secondary to systemic autoimmune disorders such as sarcoidosis, SLE, and Wegener granulomatosis. Eye specific noninfectious uveitis includes Vogt-Koyanagi-Harada syndrome, Behçet disease, birdshot retinochoroidopathy, serpiginous chorioretinitis, and ocular histoplasmosis. Posterior uveitides may involve secondary retinal vasculitis, optic neuritis, and choroiditis with granulomatous and nongranulomatous uveitic features. Posterior uveitis is believed to represent approximately 5% of all ocular inflammatory diseases, with an annual incidence of roughly 3 per 100,000 and prevalence of 30 per 100,000.
Table 9.1 Frequency of posterior uveitis
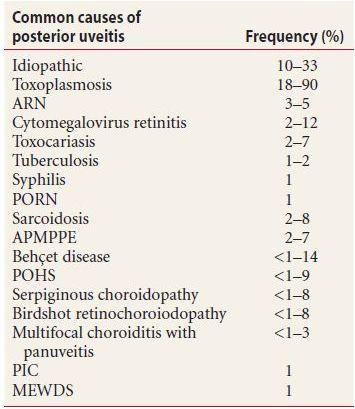
In the next section, we review posterior and diffuse uveitis management steps and some associated nuances. The final section of this chapter is dedicated to the discussion of individual disease entities.
CLINICAL MANAGEMENT OF POSTERIOR UVEITIS
When patients present with posterior uveitis, the steps for management are (a) classify the process thoroughly, (b) attempt to name the process and/or determine an etiology, (c) establish immediate control, and (d) maintain long-term control. The last two steps, however, are quite variable in the management of posterior uveitis, to the point where few generalizations are practical, and for this reason, the means for controlling posterior segment inflammation are discussed with each individual entity in the final section of this chapter.
MANAGEMENT STEP 1: CLASSIFY THE PROCESS
As delineated elsewhere, uveitis is classified as
- Acute, recurrent, or chronic in duration
- Unilateral or bilateral
- Granulomatous or nongranulomatous in character
- Anterior, intermediate, or posterior in location
It is essential that practitioners who treat uveitis are familiar with the classification scheme described in Chapter 6. The recommendations given here apply to patients with posterior uveitis (i.e., ocular inflammation with morphologic abnormalities of the retina and choroid, usually in the form of one or more white or yellow spots or amorphous patches) and diffuse or panuveitis (inflammation involving both the posterior and anterior segments).
Determining Whether Posterior Uveitis Is Acute, Recurrent, or Chronic
While patients may not know how long their posterior uveitis has been present, they may recall when their symptoms (floaters, photopsias, or visual loss) began, which is a reasonable indication of when the process started. In addition, examination findings suggesting chronicity include
- Pigmentation of the fundus lesions
- Clearly demarcated borders of the lesions
- Subretinal fibrosis associated with the lesions
- Macular edema, which, except in severe acute processes, generally suggests chronicity
These findings suggest that a patient’s disease began more than 3 months before but do not distinguish between recurrent versus chronic diseases, a determination for which we rely on a patient’s history.
Determining Whether Posterior Uveitis Is Bilateral or Unilateral
For the most part, determining whether one or both eyes have fundus lesions is straightforward. A notable subtlety, however, occurs in the patient with fundus lesions in one eye, while the fellow eye has no lesions but does in fact have vitreous cells—meaning that the patient has bilateral, though asymmetric, uveitis. This finding is common in many immune-mediated posterior segment diseases, and the distinction between unilateral and bilateral often informs our management.
Determining Whether Posterior Uveitis Is Granulomatous or Nongranulomatous
Diffuse uveitides are commonly granulomatous, especially those that are infectious. Examination findings indicating a granulomatous process include keratic precipitates or iris nodules, which may be present even with very few anterior chamber cells. Active keratic precipitates have a beige-tan color. As the process becomes inactive, these give way to pigmented clumps or take on a hyalinized glassy appearance, or resolve to leave barely visible endothelial “footprints.”
MANAGEMENT STEP 2: TRY TO ASCERTAIN THE ETIOLOGY
Visual pattern recognition is key to diagnosing posterior uveitis and diffuse uveitis that involves the posterior pole, and we feel that a straightforward approach is limited to two simple questions:
- Is the process unilateral or bilateral?
- Is the process unifocal, multifocal, or diffuse?
Applying these characteristics to Table 9.2 enables one to somewhat narrow the differential diagnosis. Eventually, as one memorizes this table, its content becomes second nature.
Table 9.2 Typical patterns of named posterior uveitides and diffuse uveitides with fundus findings
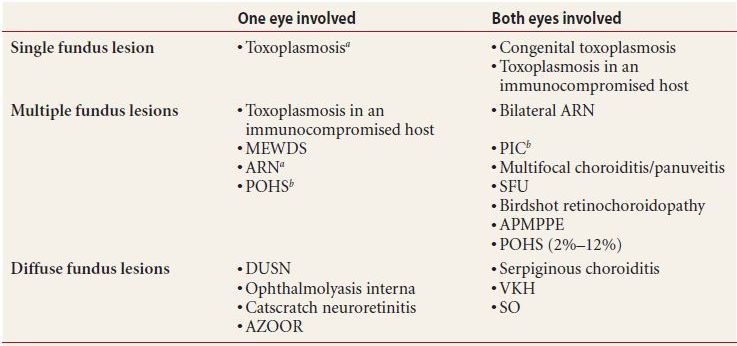
aSevere vitritis
bNo vitritis (vitritis precludes this diagnosis)
MEWDS, Multiple evanescent white dot syndrome; ARN, Acute retinal necrosis; POHS, Presumed ocular histoplasmosis syndrome; PIC, Punctate inner choroidopathy; MCP, Multifocal choroiditis/panuveitis; SFU, Subretinal fibrosis with uveitis; APMPPE, Acute posterior multifocal placoid pigment epitheliopathy; DUSN, Diffuse unilateral subacute neuroretinopathy; AZOOR, Acute zonal occult outer retinopathy; VKH, Vogt-Koyanagi-Harada syndrome; SO, Sympathetic ophthalmia.
MANAGEMENT STEP 3: TREAT FOR IMMEDIATE CONTROL
The treatment of posterior and diffuse uveitides varies greatly depending on the diagnosis, and the paradigm used in treating anterior and intermediate uveitides, that is, of establishing immediate control with—often empiric—corticosteroids before looking to measures for long-term control, is not generally applicable. For this reason, recommended treatment strategies are incorporated into the discussion of each individual entity (Tables 9.3 and 9.4).
Table 9.3 Immune-mediated posterior uveitis: common presentations
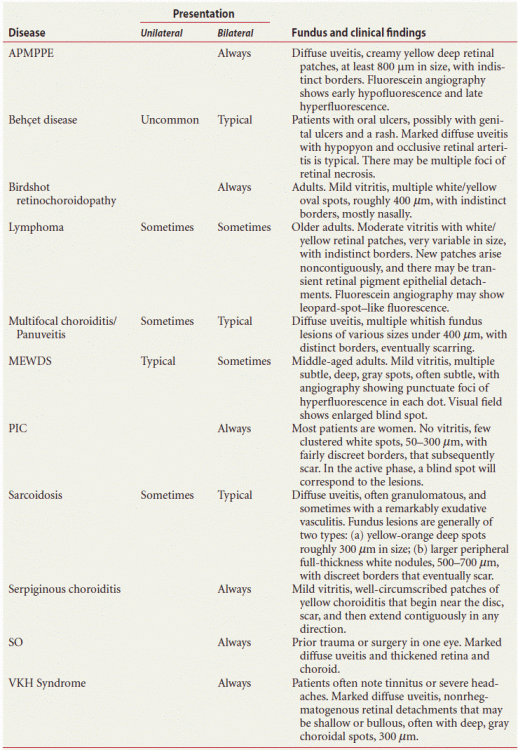
APMPPE, Acute posterior multifocal placoid pigment epitheliopathy; MEWDS, Multiple evanescent white dot syndrome; PIC, Punctate inner choroidopathy; SO, Sympathetic ophthalmia; VKH, Vogt Koyanagi Harada.
Table 9.4 Infectious posterior and diffuse uveitis: common presentations
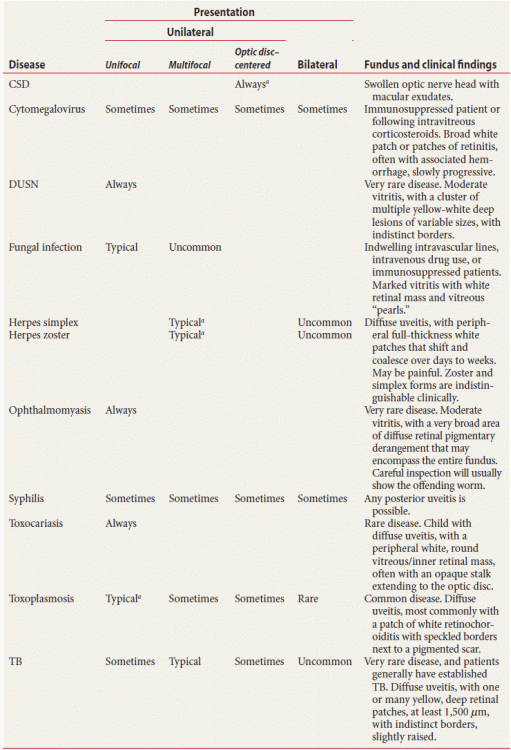
aThe optic nerve is always involved in the setting of CSD associated neuroretinitis.
CSD, Cat scratch disease; DUSN, Diffuse unilateral subacute neuroretinopathy; TB, Tuberculosis.
INFECTION-MEDIATED POSTERIOR UVEITIS AND PANUVEITIS
The diseases discussed in this section are infectious processes that are either limited to the eye or have the eye as their main site of involvement. Systemic infectious diseases that may present with posterior or diffuse uveitis are touched on briefly here, being discussed in greater detail in Chapter 15.
Acute Retinal Necrosis
First described in 1971 by Urayama, Acute retinal necrosis (ARN) is a fulminant infection of the posterior segment, caused by viruses of the herpes family. It was originally described in immunocompetent patients, although it is now well recognized in immunocompromised persons as well. The age of onset shows a bimodal distribution with peaks at 20 and 50 years. The diagnosis is based on patches of peripheral retinal necrosis (which look white and often slightly fluffy) that show rapid circumferential spread, occlusive vasculitis with arteriolar involvement, and prominent inflammation of the anterior chamber and the vitreous. Typically unilateral on presentation, involvement of the fellow eye, known as bilateral ARN or BARN, occurs in roughly 40% of cases in the absence of treatment. Retinal detachment from the necrotic area is common, complicating 75% of cases. If untreated, the disease almost invariably leads to blindness from destruction of posterior retina, optic neuropathy, optic atrophy, and retinal detachment.
Symptoms
- Floaters
- Decreased vision, occasionally with scotomata
- Red, obviously inflamed, eye
- Pain, which is often deep and scleritis-like
Clinical Findings
- Peripheral or midperipheral, white, edematous full-thickness retinal lesions.
- Lesions enlarge and coalesce rapidly into a large zone of opaque retinitis.
- The lesions progress in a circumferential configuration.
- Anterior chamber inflammation is present with significant vitritis.
- Retinal vasculitis with prominent arteriolar involvement.
- Papillitis and optic nerve edema may be present.
- Retinal detachment is unfortunately common, occuring 6 to 12 weeks after the onset of disease in the area of necrotic and nonnecrotic retina.
- After treatment, lesions are markedly atrophic with a Swiss cheese appearance, and there may be retinal pigment epithelial clumping (Figure 9.1).
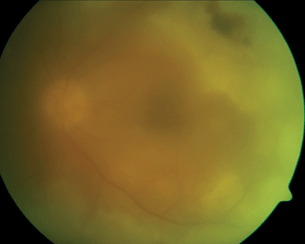
Figure 9.1 ARN with dense vitritis with extensive circumferential peripheral fluffy white retinits, retinal hemorrhage, and vasculitis.
Differential Diagnosis
- Toxoplasmosis retinochoroiditis—this can resemble ARN completely, especially in immunocompromised hosts, and is probably the most common pitfall in ARN diagnosis.
- Progressive outer retinal necrosis (PORN)—which has more posterior lesions.
- Cytomegalovirus (CMV) retinitis—which is similar but progresses much more slowly.
Diagnostic Testing
- Anterior chamber paracentesis with aqueous fluid for polymerase chain reaction (PCR)
- Diagnostic pars plana vitrectomy with vitreous fluid for PCR
- Viral particles within all layers of the infected retina on retinal biopsy
Fluorescein angiogram findings
- Peripheral area of retinal arteriolar capillary nonperfusion
- Retinal and choroidal ischemia
Treatment
- We use intravitreal injection of ganciclovir 2.0 mg or foscarnet 2.4 mg in 0.1 mL for immediate control of infection, followed by intravenous Acyclovir 1,500 mg per meter square in three divided doses for 5 to 7 days, followed by oral maintenance therapy with valacyclovir 1,000 mg orally three times per day. While we prefer this intravenous induction therapy due to the risk of herpetic encephalitis in patients with ocular involvement, some practitioners do not use IV acyclovir initially but go directly to valacyclovir oral therapy. In any case, most practitioners continue maintenance therapy, valacyclovir three times per day, indefinitely (in our experience, relapses of herpetic uveitis are common if the dose is lowered).
- Systemic (1 mg/kg/day) or periocular (20 mg subtenon) corticosteroids may be helpful 24 to 48 hours after antiviral treatment.
- Some practitioners use aspirin 650 mg daily as an anticoagulant therapy.
- Some practitioners apply laser retinopexy posterior to the area of atrophic necrotic retina for retinal detachment prophylaxis early in the recovery period. (Note: This has no effect on the spread of virus; it simply “tacks” the retina down to prevent detachment.).
Catscratch Disease (Bartonella henselae)
Catscratch disease (CSD) is a systemic infection which in some cases can involve a diffuse uveitis with neuroretinitis. CSD uveitis is recognizable as a unilateral swelling of the optic disc with macular exudates in a star-shaped configuration. There may be inner white retinal lesions in all location of the fundus with predilection for major arteries and less common retinal veins. There is a moderate cellular inflammation of the vitreous and anterior chamber. Vision loss is substantial until treatment is complete, which involves several weeks of antibiotic therapy. CSD is discussed in greater detail, including diagnosis and treatment, in Chapter 15.
Cytomegalovirus Retinitis
The cytomegalovirus (CMV) is in the herpes family of viruses. It is commonly associated with opportunistic infection in human immunodeficiency virus (HIV), acquired immune deficiency syndrome (AIDS), and immunocompromised patients. CMV retinitis is commonly the presenting illeness in patients with AIDS (upto 40% of cases). Untreated, CMV can destroy the retina within 3 to 6 months. Diagnosis is generally clinical, as serologic tests and viral culture are of limited value as majority of population show evidence of previous exposure. Roughly, one fifth of patients develop retinal detachment, especially with large areas of anterior retinal necrosis. In patients with small peripheral lesions, it can often mimic cotton-wool spots seen in HIV retinopathy. These patients can often be followed clinically with routine fundus photos to document progression in a CMV infection.
Symptoms
- Decreased vision
- Floaters
Note: CMV retinitis is painless, and the eye does not appear inflamed or irritated.
Clinical Findings
Systemic Findings
- CMV retinitis occurs in patients who are profoundly immunosuppressed, with CD4-postive lymphocyte counts less than 200 in almost all cases.
- CMV may cause esophagitis, enteritis, and a variety of other systemic manifestations separate from the occurrence of retinitis.
Ocular Findings
- Mild anterior uveitis with fine, sometimes stellate, keratic precipitates.
- Mild vitritis, invariably allowing a good fundus view.
- The retinitis can take variable appearances including; (a) fulminating necrosis, with white retina associated with hemorrhages, most commonly near the arcades in the posterior pole; (b) a “brush fire” configuration, which refers to yellow-white necrosis of the retina at the margin of previously affected, now atrophic, areas; (c) very prominent vasculitis in a frosted branch configuration, often associated with retinal hemorrhages; and (d) the indolent form: white, granular appearing retinitis without hemorrhage—common in the periphery of partially treated patients (Fig. 9.2).
- Spread is relatively slow, approximately 25 μm per day, often circumferentially, with sparing of posterior pole until late in the disease.
- There may be a primary papillitis with hemorrhage and surrounding necrotizing neuroretinitis.
- Involvement of the peripapillary region in CMV retinitis represents a 10-fold risk of encephalitis.
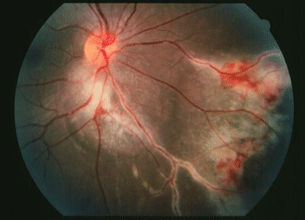
Figure 9.2 Cytomegalovirus infection with large areas of perivascular inflammation, retinitis, retinal hemorrhage, and optic nerve involvement.
Note: In evaluating and following patients with CMV retinitis, pay special attention to the borders of the active lesions, specifically the distance to the optic nerve and fovea.
Differential Diagnosis
- ARN, which progresses much (about 30-fold) more rapidly.
- PORN (even more fulminant than ARN).
- Toxoplasmosis retinochoroiditis—which may be quite similar, although is generally more limited in distribution within the retina than CMV, and has less of a predilection for markedly immunosuppressed patients.
- Pneumocystis carinii choroiditis—these spots are deeper and more yellowish, not presenting as a frank retinitis.
Diagnostic Testing
The diagnosis is usually made clinically, and ancillary tests are not necessary. Aqueous humor or vitreous samples can be tested with PCR if the diagnosis is doubtful. Serologic testing for anti-CMV antibodies could confirm prior exposure to this infection but is generally not helpful in establishing the diagnosis.
Treatment
- Our preferred approach is to administer intravitreal ganciclovir 2.0 mg or foscarnet 2.4 mg in 0.1 mL and immediately initiate maintenance therapy with oral valganciclovir 900 mg PO two times per day. We see patients 1 week later, by which point the retinitis should have improved dramatically. Fundus photography is the optimal way of following disease progression. In cases with severe disease, repeat weekly injections may be necessary to clear the retinitis. Once the infection has been controlled, we see patients every 3 months while on maintenance therapy and every 6 months or yearly once immune function has been restored.
- Intravenous ganciclovir 5 mg per kg every 12 hours for 2 weeks may be used in lieu of intravitreal injection, followed by oral ganciclovir maintenance therapy.
- Intravenous foscarnet or cidofovir is used in cases of ganciclovir resistance.
- The ganciclovir intraocular implant is a means of providing lasting control if the infection is known to respond to this agent (which is usually—but not always—the case). It does not suppress systemic infection. Note: resistance to ganciclovir is common, with 20% or more patients experiencing relapse during maintenance therapy.
- Restoration of immune system function with antiretroviral therapy will result in durable control of CMV infection, and the incidence of CMV plummeted after the introduction of highly active antiretroviral therapy (HAART) in the late 1990s.
Diffuse Unilateral Subacute Neuroretinitis (Unilateral Wipe Out Syndrome)
Diffuse unilateral subacute neuroretinitis (DUSN) is a rare infectious retinochoroiditis associated with subretinal nematode, Toxocara canis and Baylisascaris procyonis. This clinical entity was first described by Gass and Scelfo in 1978. The smaller worm T. Canis, a common hookworm of parasites of dogs, is 400 to 1,000 μm in size. The larger worm B. procyonis is an intestinal round worm of raccoons and squirrels and is 1,500 to 2,000 μm in length. The worm propels itself by a series of slow coiling and uncoiling movements. It is likely that a variety of nematodes can stimulate the inflammatory response typical of this disease. Early in the disease, patients notice floaters, mild visual loss, and, occasionally, migratory paracentral or central scotoma. An afferent papillary defect is usually present. If untreated for an extended period of time, late manifestations include a dense central scotoma and eventually severe vision loss. Stool samples for ova and parasites are unrevealing, and serum eosinophilia is not found. The live nematodes can live up to 4 years or longer in the subretinal space.
Clinical Findings
- One should be suspicious of a worm if there is marked retinal destruction in only one eye, often in the form of recurrent crops of multiple focal gray or yellow subretinal lesions in the outer retina, with retinal pigment epithelium (RPE) loss in a linear configuration. There may be local serous detachments with slight disc edema, and the vitreitis is not severe.
- An afferent papillary defect should be present in severe cases.
- A visible subretinal worm can be seen in less than 50% of cases (Fig. 9.3).
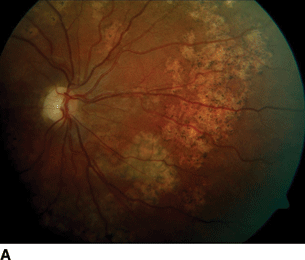
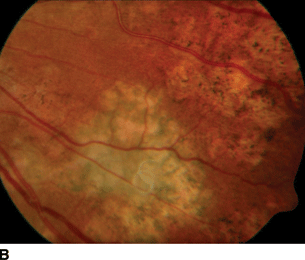
Figure 9.3 A, B: DUSN with diffuse retina pigment epithelium changes with large areas of atrophy and focal area of retinal whitening. The worm is in the subretinal space with overlying retinal whitening, vitritis, and retinal edema.
Differential Diagnosis
- A visible worm obviously establishes the diagnosis.
- Early DUSN can be mimicked by optic neuropathies involving disc edema.
- Toxoplasmosis may present with unilateral retinal destruction, but the degree of vitritis would generally be greater and the retinal destruction more complete.
- Herpetic retinitis, like toxoplasmosis, might also present with unilateral retinal destruction, though with more vitritis and more complete retinal destruction.
- CMV could also present with marked retinal wipeout in one eye, but the patient’s history of immunosuppression should be easily recognizable.
Diagnostic Testing
Fluorescein angiogram
- Leakage from the capillaries on the optic nerve head.
- Patchy area of early choroidal filling with delay in retinal arterioles.
- Secondary choroidal neovascular membrane can be detected.
Electroretinogram
- Normal at early stage
- Moderate to severe decrease in Electroretinogram (ERG) amplitude, B wave more affected than A wave
Treatment
- Laser photocoagulation to kill the subretinal worm is the treatment of choice. One should anticipate a marked inflammatory response, and we treat patients with corticosteroids (prednisone 1 mg/kg for 1 week) following laser therapy.
- Thiabendazole 22 mg/kg PO twice a day for 4 days may kill the worm and has the obvious advantage of killing worms that cannot be seen.
- Ivermetin was used successfully in a small number of patients with DUSN.
Ophthalmomyiasis
Humans can become infected with the larvae of a number of parasitic organisms that have life cycles requiring mammalian hosts, and ophthalmomyiasis is the condition of having such an infection with a fly larva, or maggot, invading the eye. These larvae appear to land on the ocular surface via airborne travel or when deposited by flies (the order Diptera, including flies that live around livestock, is the main culprit). Infection of the ocular surface is termed ophthalmomyiasis externa. These patients present with red, irritated eyes, and examination shows a fly larva (approaching 1 mm in size) in the cornea or conjunctiva, from where removal of the larva is curative. Ophthalmomyiasis interna results if the larva penetrates the sclera to reach the anterior chamber, vitreous, or subretinal space. The organism dies in the vitreous, inciting a marked localized vitreitis or dense endophthalmitis. In the subretinal space, however, the organisms can live and actually grow to exceed 3 mm in length, while moving around the fundus, leaving pathognomonic linear tracks in the RPE.
Clinical Findings
- Mild anterior uveitis or vitreitis
- Diffuse alteration of RPE with numerous linear atrophic tracks
- Subretinal hemorrhage
- Retinal detachment
- Optic atrophy is a late finding
Differential Diagnosis
The finding of linear tracks in the RPE is unique to this condition, and this presentation does not present a diagnostic dilemma. A unilateral localized or diffuse vitreitis presents far more diagnostic possibilities, including
- Toxoplasmosis
- Syphilis
- Toxocariasis
- Herpetic retinitis
Diagnostic Testing
Fluorescein angiogram
- Extensive area of linear hyperfluorescence due to window defect of RPE
- Mild area of leakage and staining in the area of the larvae
Treatment
- Laser photocoagulation of the subretinal larvae—which will incite a marked inflammatory reaction.
- Pars plana vitrectomy for the removal of vitreous larvae.
- Medical treatment to kill the larva is not achievable.
Pneumocystis Choroiditis
Pneumocystis jiroveci (PJ, formerly called Pneumocystis carinii) is an indolent organism commonly associated with advance HIV patients and opportunistic infections. PJ choroiditis is now an uncommon ocular presentation due to prophylactic treatment with trimethoprim/sulfamethoxazole. Extensive ocular disease was common during the earlier era of HIV. This form of uveitis does not cause visual loss and is most significant as a marker of a potentially fatal infection than as an eye disease.
Clinical Findings
Systemic Findings
- Patients with PJ choroiditis are generally profoundly immunosuppressed, with multiple concomitant infections.
Ocular Findings
- Both eyes generally show multiple scattered pale yellow choroidal lesions 300 to 3,000 μm in size with hazy borders
- There is minimal anterior chamber and vitreous inflammation
- The lesions enlarge slowly over months with little effect on visual acuity
- Pigmentary in the area of subretinal lesions
Differential Diagnosis
- Acute Posterior Multifocal Placoid Pigmentary Epitheliopathy (APMPPE)
- Lyme disease
- Syphilis
Diagnostic Testing
- Fluorescein angiogram shows early hypofluorescence and late hyperfluorescence, staining
- Presence of systemic Pneumocystis infection in an immunocompromised patient
Treatment
- 3 weeks’ regimen of IV trimethoprim (20 mg/kg body wt/day) and sulfamethoxazole (100 mg/kg body wt/day) or pentamidine (4 mg/kg body wt/day)
- Trimethoprim 160 mg/sulfamethoxazole 800 mg PO twice daily
Presumed Ocular Histoplasmosis Syndrome
Presumed ocular histoplasmosis syndrome (POHS) is a noninfectious immunologic response to the fungus Histoplasma capsulatum. The organism is found worldwide and is endemic to the Midwestern states of the United States. The organism is found in soil and is readily inhaled. The lesions are commonly found in the lungs with old calcific lesions on chest X-rays. The disease is characterized by the classic triad of punched out peripheral chorioretinal lesions (“histo spots”), peripapillary atrophy, and an asymmetric maculopathy from the presence of choroidal neovascular membrane are the typical features on examination. Classic findings of ocular histoplasmosis can be seen in 1.6% to 12.9% of the population in endemic areas. The disease will most commonly occur during the third and fourth decade with males more likely to present with bilateral macular involvement. In patients without macular lesions, the risk of choroidal neovascularization (CNV) is roughly 2%, while the risk of CNV is 25% in patients with macular lesions. Peripapillary changes in histoplasmosis occur due to underlying choroiditis with a line of pigment bordering the optic disc.
Clinical Findings
- Histo spots are 0.2 to 0.7 disc diameter in size, depigmented, and may have a central area of pigment.
- The periphery has 10 to 20 histo spots posterior to the equator, appearing during the teen years.
- Peripheral streak lesions (probably aggregation of spots).
- Macular lesions develop after the second decade with abrupt change in vision.
- No vitreous cells. In fact the presence of vitreous cells disqualifies this diagnosis.
- Choroidal neovascular membrane in the area of maculopathy develops.
Note: Histoplasma skin testing may activate the disease and should be avoided.
Differential Diagnosis
- Angioid streaks
- Choroidal rupture
- Punctate inner choroidopathy (PIC)
- Pathological myopia
- Idiopathic CNV
Diagnostic Testing
- The diagnosis is clinical, but a chest radiograph to rule out pulmonary histoplasmosis lesions may benefit the patient.
Treatment
Therapy is directed at the choroidal neovascular membranes which complicate this condition. It is not needed for the histoplasma organisms themselves.
- Focal laser photocoagulation for juxtafoveal and extrafoveal lesions.
- Photodynamic therapy with verteporfin for subfoveal lesions.
- Antivascular endothelial growth factor (Anti-VEGF) therapy.
- Submacular surgery has been used to remove neovascular membranes, with varying degrees of success.
Progressive Outer Retinal Necrosis
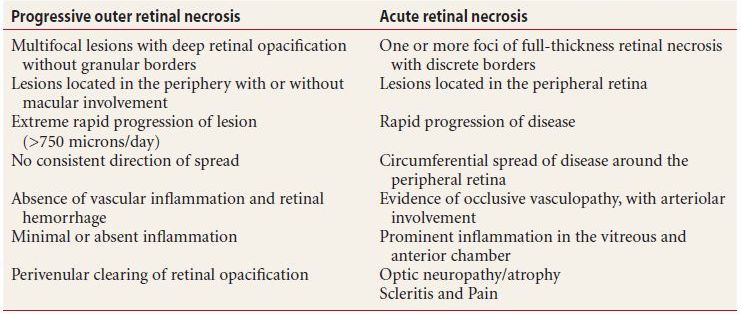
In 1987, Jabs reported three cases of varicella-zoster virus retinitis in immunocompromised patients. Forster later described two additional cases and coined the term “progressive outer retinal necrosis.” PORN now is recognized as the second most common retinal infection associated with the herpes family of viruses in AIDS patients with CD4 lymphocyte counts less than 100 cells/mL, with a median of 21. There may be a recent history of herpes zoster ophthalmicus ipsilaterally. Patients often present with painless decrease in vision or loss of visual fields. Presenting visual acuity can range from 20/20 to no light perception (NLP). The involved eye shows no injection or external signs of infection. Retinal vascular inflammation is absent and optic nerve involvement is rare. Rapid vision loss is a feature of the disease with progression to NLP in weeks. See Table 9.5.
Table 9.5 Diagnostic criteria progressive outer retinal necrosis vs. acute retinal necrosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


