7
Anterior Uveitis
Raluca Raducu and I. Willard Abrahams
Anterior uveitis (AU) is synonymous with iritis and is defined by the presence of cells or cellular aggregates visible in the anterior chamber using slit-lamp biomicroscopy. It is the most common form of intraocular inflammation. This chapter begins by discussing the clinical management of AU. Diseases that cause iritis are listed and discussed at the end of this chapter.
CLINICAL MANAGEMENT OF ANTERIOR UVEITIS
The steps for managing AU are (a) classify the process, (b) attempt to determine an etiology, (c) establish immediate control, and (d) establish long-term control. (See Table 7.1.)
Table 7.1 Examination findings to assist in classifying anterior uveitis

Note: conjunctival nodules merit biopsy with suspicion of sarcoidosis
Management Step 1: Classify The Process
In managing all patients with uveitis, we find it most helpful to organize our thoughts by classifying the process in four respects:
- Acute, recurrent, or chronic in duration
- Unilateral or bilateral
- Granulomatous or nongranulomatous in character
- Anterior, intermediate, or posterior in location
In addition, we include any relevant associated clinical contexts in this schema, such as a systemic disease known or presumed to be the cause of the patient’s iritis. (See the uveitis classification scheme explained in Chapter 6.)
The present discussion is oriented toward the situation in which a patient’s ocular inflammation has been determined to be anterior, meaning that item 4 in the above list has already been clarified.
Determining Whether Anterior Uveitis Is Acute, Recurrent, or Chronic
Most patients can recall when their iritis started, so whether the process is acute, recurrent or chronic is often straightforward simply by taking the history. It gets tricky, however, when the patient recalls having had similar iritis like symptoms in the same or the fellow eye at some point in the remote past but was not diagnosed at that time. Can we say the current episode is a recurrence, based on that bit of history?
Fortunately, iritis usually leaves clues behind, which a careful examination can detect. Examination findings suggesting that a patient has had a prior episode of iritis are listed in Table 7.1 and include (a) remnants of keratic precipitates (KP); (b) pigment clumps on the anterior lens face; (c) posterior synechiae; (d) anterior synechiae, which might only be visible on gonioscopy and even then might not be true synechiae but rather a “dirty” or scarred look to the inferior anterior chamber angle; and (e) residual anterior vitreous cells that “spilled over” during a prior inflammatory episode. We document these findings or the lack thereof if there is any question of a prior inflammatory episode in either eye.
Another difficulty in determining disease chronicity arises when a patient cannot recall or was not aware of when his or her iritis started, usually because it is asymptomatic. Here again, the examination can usually answer the question. Examination findings suggesting that iritis has been longstanding include (a) band keratopathy and (b) loss of contour to the iris face, often with a washout of the iris color. Posterior synechiae, even many clock hours of them, are unfortunately not helpful, since they can form very quickly in very severe iritis and so do not indicate a long-standing process, unless occlusio pupillae (a fibrovascular membrane in the papillary space) has developed, which is, however, rare in developed countries.
Determining Whether Anterior Uveitis Is Bilateral or Unilateral
This question is also usually quite straightforward based on history and examination. The only common pitfall is the patient who has acute iritis in one eye and therefore appears to have a unilateral process but who has in fact had iritis in the other eye previously and thus actually has a bilateral, recurrent process. Examination findings listed above and in Table 7.1 can help one determine if the currently uninflamed eye has been inflamed in the past.
Determining Whether Anterior Uveitis Is Granulomatous
This finding is made entirely by examination. We look for KP or the remnants thereof and iris nodules, which may be at the iris margin, on the face of the iris stroma, or in the anterior chamber angle (seen on gonioscopy). A single nodule or KP is sufficient to declare the process granulomatous. It is worth noting that we make a careful search for conjunctival nodules to biopsy in patients with iritis, since a biopsy showing noncaseating granulomas generally establishes the diagnosis of sarcoidosis, regardless of whether the iritis appears granulomatous.
Management Step 2: Try to Ascertain The Etiology
The most useful information indicating that a patient may have an underlying disease associated with his or her AU is gleaned during the clinical encounter, that is, from the history, review of systems, and examination. Laboratory or radiographic ancillary testing can be helpful in some settings.
With respect to etiology, we find it helpful to think of AU in four categories:
- Iritis syndromes which are diagnosed entirely on the basis of certain highly characteristic examination findings.
- Iritis secondary to other eye problems. These are also diagnosed solely by the examination findings.
- Iritis secondary to underlying systemic conditions. These diagnoses are suggested by the history and review of systems and supported by ancillary testing. The wide variety of laboratory and radiologic tests that many practitioners associate with uveitis are relevant to this disease category.
- Iritis for which no etiology is apparent based on the history, review of systems, or examination or for which a suspected etiology was not borne out by ancillary testing. Roughly half of all AU falls in this group, having been excluded elsewhere
- Iritis secondary to underlying systemic conditions. These diagnoses are suggested by the history and review of systems and supported by ancillary testing. The wide variety of laboratory and radiologic tests that many practitioners associate with uveitis are relevant to this disease category.
For a patient with AU, the process of ascertaining an etiology thus involves working sequentially through the first three of these categories, with the fourth category being the repository for undiagnosed disease.
Category 1: Iritis Syndromes
In certain cases, a patient’s iritis is recognizable as a syndrome diagnosed entirely on the basis of highly characteristic examination findings. No supporting laboratory tests are needed for these diagnoses, which are listed in Table 7.2 and include
Table 7.2 Anterior uveitis syndromes
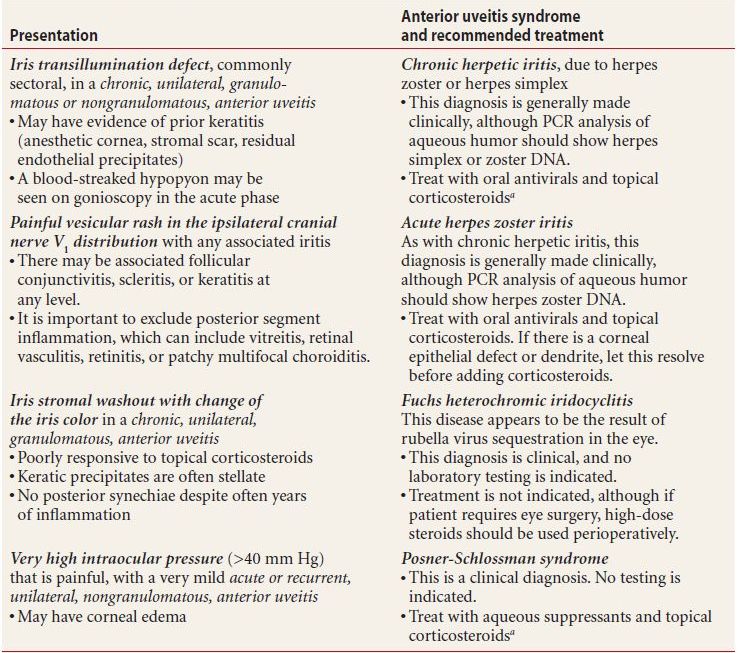
a “Topical corticosteroids” refers to prednisolone 1% or dexamethasone 0.1% eyedrops.
PCR, polymerase chain reaction.
- Chronic herpetic uveitis, indicated by an iris transillumination defect
- Acute herpes zoster uveitis, indicated by a painful vesicular rash in the ipsilateral cranial nerve V1 distribution
- Posner-Schlossman syndrome, or glaucomatocyclitic crisis, indicated by a very high intraocular pressure (IOP), mydriasis, and mild, acute, unilateral, nongranulomatous iritis
- Fuchs heterochromic iridocyclitis, the classic presentation being stellate KP and washout of the anterior iris face, usually without synechiae or other sequelae typical of chronic iritis
- Posner-Schlossman syndrome, or glaucomatocyclitic crisis, indicated by a very high intraocular pressure (IOP), mydriasis, and mild, acute, unilateral, nongranulomatous iritis
Category 2: Iritis Secondary to Other Eye Problems
Iritis is occasionally the most prominent anterior segment finding associated with other problems in the eye. Usually, these problems are quite obvious from either the history or the examination. Common examples are listed in Table 7.3 and include
Table 7.3 Anterior uveitis due to other problems in the eye

- Iritis due to ocular ischemic syndrome
- Lens-associated iritis due to mature cataract
- Lens-associated iritis due to trauma
- Traumatic iritis without lens involvement
- Iritis due to a tight scleral buckle
- Iritis due to chronic retinal detachment
- Iritis due to intraocular lens malpositioning
Category 3: Iritis Secondary to Underlying Systemic Conditions
A number of diseases present initially with iritis, and it is incumbent upon the treating practitioner to rule these out. As noted above, the history, review of systems, and examination will reveal by far the most useful information along these lines. While additional information can be obtained from laboratory and imaging studies, we discourage a “shotgun” approach to testing, whereby one orders a wide range of tests and then struggles to find some meaning in the confusing mix of results, only some of which are occasionally relevant, the rest being confusing at best and misleading at worst. While the variety of presentations is limitless, and it is thus probably impossible to list the ancillary diagnostic tests that are indicated for each and every case of iritis, we recommend the following general guidelines for ancillary testing:
- Begin with the assumption that no tests will be ordered.
- Review and be certain that the presentation does not match that of an AU syndrome (as described above and in Table 7.2) and that no other eye problem (listed above and in Table 7.3) explains the inflammation.
- Order a workup for any patient with any of the following highly suggestive examination findings:
- nongranulomatous iritis
- hypopyon
- granulomatous anterior iritis
This is further explained in Table 7.4.
Table 7.4 Highly suggestive examination findings and the appropriate ancillary workup
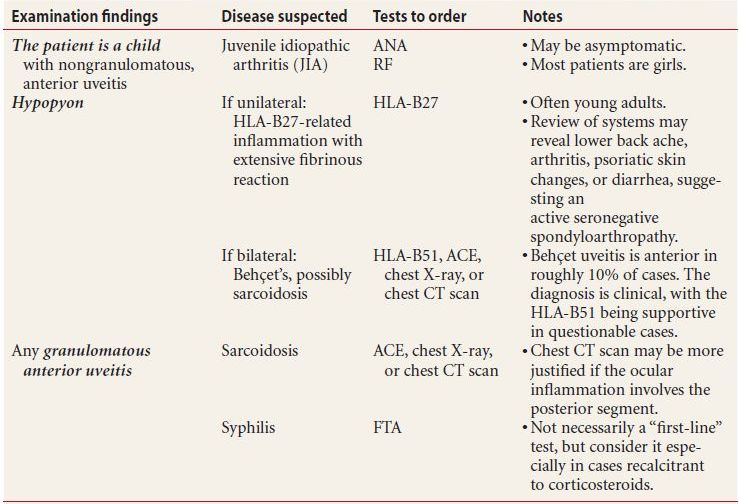
HLA, human leukocyte antigen; PCR, polymerase chain reaction; ACE, angiotensin-converting enzyme; ANA, antinuclear antigen; RF, rheumatoid factor; FTA, fluorescent treponemal antigen; CT, computed tomography.
- Order a workup for any patient with notable findings on history or review of systems, and include (the associated diseases are in parentheses)
- Chronic low back pain in young adults (ankylosing spondylitis [AS])
- Psoriasis with arthritis (psoriatic arthritis)
- Oral ulcers and/or genital ulcers (Behçet disease)
- Pulmonary symptoms or any raised skin lesions (sarcoidosis)
- Red, swollen, painful ears, nose, or throat (relapsing polychondritis)
- Chronic diarrhea (more than three loose bowel movements per day, lasting more than 3 months, suggesting inflammatory bowel disease)
- A history of sexually transmitted disease (syphilis)
- Anesthetic macular or nodular skin lesions (leprosy)
- A child with fever, rashes, and/or tongue swelling (Kawasaki disease)
- Chronic low back pain in young adults (ankylosing spondylitis [AS])
Suspect diseases and the appropriate workup are explained in Table 7.5.
Table 7.5 Significant findings on history and review of systems, and appropriate tests to order
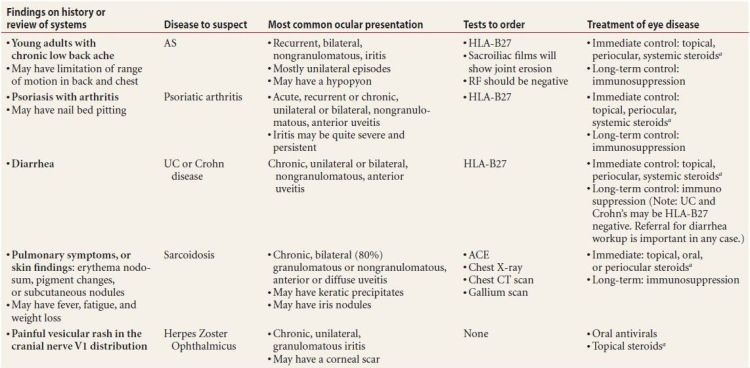

aSteroids in this table refers to corticosteroids. “Topical steroids” refers to prednisolone 1% or dexamethasone 0.1% eyedrops.
ANA, antinuclear antibody; RF, rheumatoid factor; ACE, angiotensin-converting enzyme; FTA, fluorescent treponemal antigen; VDRL, venereal disease research laboratory; ESR, erythrocyte sedimentation rate; HLA, human leukocyte antigen.
- Order a workup for any patient whose iritis has persisted undiagnosed for a year or has recurred after initially resolving. The reason here is that some of the diseases that cause chronic AU will show unremarkable laboratory test results initially, but these tests will convert to positive over time if the iritis persists. This is particularly true in three situations:
- Sarcoidosis, in which the uveitis can take virtually any manifestation. We, therefore, often repeat the ancillary tests for this (angiotensin-converting enzyme and chest X-ray or CT scan) roughly on a yearly basis as long as the iritis persists or if it recurs.
- Juvenile idiopathic arthritis, in which the antinuclear antibody (ANA) may become positive over time. For this reason, we repeat the ANA yearly in any child with undiagnosed nongranulomatous iritis.
- Syphilis. We might not order tests for syphilis when a patient presents initially, but we have a low threshold for ordering a fluorescent treponemal antigen (FTA) for a patient whose iritis persists with no apparent association, particularly if the condition seems incompletely responsive to corticosteroid therapy. One must be careful interpreting positive syphilis tests in the setting of iritis, however, such a result does not necessarily mean that the patient’s eye disease is syphilis related. For this reason, we reserve syphilis testing for situations in which we genuinely suspect that an iritis is infectious.
Management Step 3: Treat for Immediate Control
With very few exceptions, AU can and should be treated with topical corticosteroids. In the United States, the most popular eyedrop for this purpose is prednisolone acetate 1%, although dexamethasone 0.1% or prednisolone sodium phosphate 1% are reasonable alternatives. We do not recommend using other medications or lower concentrations of these medications. The only possible exception is difluprednate 0.05%, which is a much newer corticosteroid eyedrop that appears to belong among the potent corticosteroid agents just mentioned. Its optimal dosing has not been firmly established, so we will not include it in this discussion, acknowledging that it may play a role in iritis therapy eventually.
Standard Treatment Regimen for Achieving Immediate Control in Most Forms of Iritis
- Begin with hourly corticosteroid eyedrops (as named above) while awake—usually at least 14 drops per day, and use these for at least 1 week. We generally do not schedule follow-up within that week, since it will be too soon to make any useful therapeutic decisions.
In follow-up at 1 week, the number of anterior chamber cells/high-power field (hpf) should be half of what it was at initial presentation. If it is not, then suspect that the patient is not using the eyedrops or that the condition may be infectious—typically herpetic (see “special cases,” discussed below).
Note: We do not recommend using corticosteroid eyedrops any less frequently than every hour in the initial treatment of any iritis. While this approach may work in some cases, very commonly it is simply not enough, and patients end up needing to use these eyedrops for much longer than would have been necessary had they been treated more frequently initially.
- Continue hourly therapy with weekly visits until there are five or fewer cells/hpf. This may mean a few weeks of hourly therapy, with assessment each week anticipating that the number of anterior chamber cells/hpf will decrease by half at each visit.
- Once there are five or fewer cells/hpf in the anterior chamber, reduce the dose frequency to every 2 hours. Reevaluate every 2 weeks, and as long as the inflammation is not increasing, decrease the corticosteroids to every 2 hours for 2 weeks, four times per day for 2 weeks, three times per day for 2 weeks, two times per day for 2 weeks, one time per day for 2 weeks, and then discontinue the therapy. Thus, the increments in our standard eyedrop taper are: every 1 hour, every 2 hours, four times per day, three times per day, two times per day, one time per day, and then stopping the drug.
- If the disease flares during this period, one needs to consider using either a slower taper or periocular corticosteroid injections, as described below.
- Use cycloplegia, typically homatropine 5% twice daily, until there are fewer than five cells/hpf in the anterior chamber and then discontinue it.
Treatment Regimens for Immediate Control in Special Cases of Iritis
- Hypopyon iritis. This condition, which usually indicates that the patient carries the HLA-B27 gene, can require many weeks of hourly eyedrop therapy before being amenable to taper. It is important to verify that the uveitis is truly an anterior process. Assuming it is, we recommend two modifications to the standard treatment regimen described above.
Firstly, in addition to hourly corticosteroid eyedrops, we often prescribe oral corticosteroids, equivalent to prednisone 1mg/kg/day, continuing these for the first 7 days, or we use periocular injections as an alternative to oral corticosteroids. The injectable options are dexamethasone 2 mg, 0.5 mL, which will leave the eye in roughly 24 hours, or betamethasone phosphate/acetate 3 mg, 0.5 mL, which will leave the eye in roughly 10 days. Patients should use their eyedrops as per the standard regimen if either of these is used. If the patient has taken corticosteroids before and is not known to be a brisk corticosteroid-IOP responder, we often inject triamcinolone acetonide 20 mg, 0.5 mL, in the subtenon space. This will remain around the eye for several months, so that the patient can use corticosteroid eyedrops four times per day instead of hourly. (This approach is also advantageous for patients who are unable to use hourly eyedrops.)
Secondly, we taper the corticosteroid eyedrops by one increment each month, rather than every 2 weeks, since very aggressive iritis is well known to recur if a very slow taper is not used.
- Herpetic iritis. If we suspect this, usually because the patient either has an iris transillumination defect, has a blood-streaked hypopyon, or has responded poorly to corticosteroid therapy, we add oral antivirals to the standard regimen described above. Acyclovir 400 mg (800 mg for zoster) five times per day, valacyclovir 500 mg (1,000 mg for zoster) three times per day, or famciclovir 500 mg three times per day are all acceptable and appear to be equally efficacious. Once the patient is using corticosteroid eyedrops three times per day, we decrease the antiviral coverage to two times per day for acyclovir or one time per day for valacyclovir or famciclovir.
- Fuchs heterochromic iridocyclitis. This is the only form of iritis that we do not treat once the diagnosis is established, since treatment effects no discernible change in the inflammation. In many cases, the initial presentation of Fuchs’ is not entirely typical, and it is not unusual that the diagnosis is made as one of exclusion after attempts to treat with corticosteroids and often with antivirals as well.
- Iritis flares during taper. In these cases, the options are a periocular triamcinolone injection, typically with 20 mg or less, depending on the degree of inflammation, or simply a slower taper of corticosteroid eyedrops, decreasing by one increment each month rather than every 2 weeks.
Management Step 4: Treat for Long-Term Control
We consider long-term control to be the process of keeping a patient’s eyes inflammation free after the initial inflammatory episode has been treated. This is by far the most challenging aspect of uveitis therapy.
Certainly, in many cases, a patient’s iritis may not recur after the initial treatment is tapered; the disease is quiescent, and no long-term control therapy is required. Frequently, however, the iritis recurs either during the initial corticosteroid taper or within 3 months of stopping it; this disease is chronic, and long-term control measures are needed.
The two general approaches to long-term control of AU are corticosteroids and immunosuppressives.
Corticosteroids
Corticosteroids are an acceptable means of maintaining long-term control of iritis if (a) the dose required is low and likely to be well tolerated and (b) the patient’s iritis is not believed to be associated with a systemic disease.
We occasionally use corticosteroid eyedrops long-term if complete control of iritis is maintained with three or fewer drops per day (one drop three times per day), a dose that many patients can take for several months before developing side effects. It is important to be certain that the inflammation is indeed controlled when using corticosteroid eyedrops for a longterm, since the worst mistake a clinician could make, we feel, is to use these drops for a longterm and still tolerate inflammation (we generally do not tolerate more than two cells/hpf), thereby subjecting a patient to the undesirable effects of both disease and drugs.
- Periocular corticosteroid injections are an effective means of maintaining long-term control in some patients with iritis, provided they are not needed too frequently—which to us means one triamcinolone 20 mg injection roughly every 6 months. This approach is helpful in enabling patients to avoid the use of corticosteroid eyedrops when they would otherwise require these agents four times per day or more.
IOP elevation is common when corticosteroids are used for a long time in any form, and IOP-lowering eyedrops are frequently required as adjunct therapy.
Not uncommonly, patients with chronic uveitis will not tolerate corticosteroids, because of intractable IOP elevation or posterior subcapsular cataract. In this case, we resort to immunomodulatory therapy.
- We use immunomodulatory therapy for AU in two general circumstances: (a) when patients cannot tolerate corticosteroids, usually due to IOP elevation but occasionally due to early cataract formation; (b) iritis in the setting of a systemic inflammatory disease. We discuss the use of these agents further in Chapter 19.
DISEASES THAT CAUSE ANTERIOR UVEITIS
Autoimmune Causes: HLA-B27-Associated AU
- The HLA-B27 gene is located on the short arm of chromosome 6.
- It is present in 1% to 8% of the general population, varying by race.
- The associated uveitis affects males more than females.
- The AU is often severe with a fibrinoid hypopyon, with a recurrent course, eventually involving both eyes, though usually not simultaneously.
- HLA-B27 diseases are termed seronegative spondyloarthropathies (“seronegative” referring to a negative rheumatoid factor). They include
- Ankylosing spondylitis
- Reiter syndrome (reactive arthritis)
- Inflammatory bowel disease
- Psoriatic arthritis
- Postinfectious or reactive arthritis
Ankylosing Spondylitis–Associated AU
Incidence
- Patients with AS usually have a positive family history; males are more frequently affected (80% males), and 88% are positive for HLA-B27.
- There is a 25% chance that an HLA-B27-positive patient with AS will develop AU.
Clinical Findings
- Women often show minimal back symptoms
- Low back pain more than 3 months unrelieved by and often worse after rest
- Decreased lumbar motion as the disease progresses
- Aortitis in 5% of patients, other patients developing pulmonary apical fibrosis and chest wall pain or stiffness
Diagnostic Testing
- The HLA-B27 test establishes the presence of the gene.
- CT scan or X-ray will demonstrate changes of the lumbosacral spine or pubic symphysis.
Treatment
- Physical therapy to help preserve spinal and neck mobility
- Oral NSAIDS
- Oral corticosteroids or immunomodulatory agents including methotrexate or tumor necrosis factor alpha antagonists
Reiter Syndrome–Associated AU
The diagnostic triad for Reiter syndrome consists of urethritis, conjunctivitis, and arthritis. The condition occurs most frequently in young adult males (90%), and 85% to 95% are HLA-B27 positive. Reiter syndrome may be triggered by episodes of urethritis caused by Chlamydia or Ureaplasma urealyticum or by diarrhea or dysentery caused by Campylobacter, Shigella, Salmonella, and Yersinia. Arthritis starts within 30 days of infection in 80% of patients.
Clinical Findings
Ocular findings
- Early on, may have a mucopurulent papillary conjunctivitis
- Severe chronic recurrent acute attacks of iritis (50%)
- Heavy flare, early synechiae
- Subepithelial keratitis may leave scars
Systemic findings
- Recurrent asymmetric lower extremity migratory polyarthritis
- Sacroiliitis (1/3)
- Keratoderma blennorrhagica of hands and feet (looks like pustular psoriasis) (Fig. 7.1)
- Nail bed pitting
- Palatal and tongue aphthous ulcers
- Enthesitis, fasciitis, achilles tendonitis
- Achilles tendonitis
- Nonspecific urethritis, prostatic fluid culture negative
- Often after epidemic dysentery or STD from Chlamydia, Ureaplasma, Shigella, Salmonella, and Yersinia
- Circinate balanitis of the distal penis: persistent, scaly, erythematous, circumferential rash
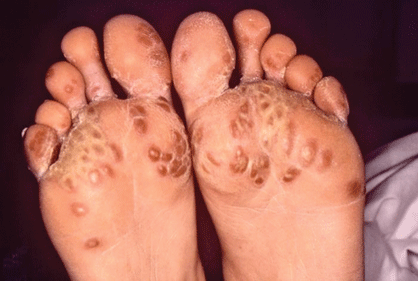
Figure 7.1 A patient with Reiter syndrome with keratoderma blennorrhagia of his feet. The lesions are dry, scaly, and hyperpigmented.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


