10
Retinal Vasculitis
John J. Huang
OVERVIEW
Retinal vasculitis represents a heterogeneous group of disorders with retinal vascular inflammation and associated ocular inflammation. Many of the patients with retinal vasculitis have been previously diagnosed with an associated systemic disorder, but some may develop retinal vasculitis as the initial manifestation of an underlying systemic autoimmune or infectious disorder. Less commonly, retinal vasculitis may occur as an isolated disease (Table 10.1). The critical role of the ophthalmologist diagnosing retinal vasculitis is to determine—or at least weigh in on—the likelihood of associated systemic and central nervous system inflammation because the underlying etiology of the vasculitis is among the most important factors informing therapy.
Table 10.1 Disorders associated with retinal vasculitis

CLINICAL MANIFESTATIONS
Patients with retinal vasculitis most often report a painless loss of vision with symptoms of floaters. If the retinal vascular changes are in the periphery, the patient may be asymptomatic. The involved area of the retina and the retinal vessels may demonstrate a relative or an absolute scotoma on visual field testing. Common clinical manifestations of retinal vasculitis include vascular sheathing, vitreitis, intraretinal hemorrhage, macular edema, optic nerve edema, and vascular leakage (Fig. 10.1).
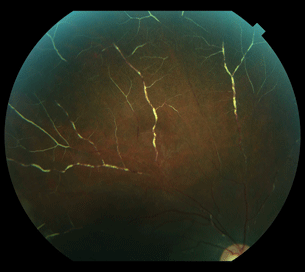
Figure 10.1 Occlusive vasculitis with complete sclerosis of the peripheral retinal arteries and veins.
DIAGNOSTIC TESTING
Fluorescein Angiography
Retinal vasculitis is generally best demonstrated by fluorescein angiography (FA). Vascular leakage is the hallmark of this disorder, involving the arteries (and termed arteritis) or veins (termed phlebitis). There may be areas of vascular occlusion with capillary dropout, and macular edema, optic disc leakage or vascular sclerosis may be evident (Fig. 10.2). Macular ischemia may result in visual loss that is irreversible even after the inflammatory process has resolved. FA also indicates whether the inflammation involves veins, arteries, or both. FA is also useful for gauging the efficacy of systemic therapy for patients under treatment; inflammation that is not apparent on clinical examination may be evident on FA. Such patients generally undergo at least 2 angiograms yearly to follow their disease course.
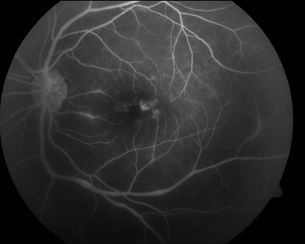
Figure 10.2 Diffuse vasculitis with an extensive inflammation of the retinal veins with evidence of mild CME.
Thorough Ocular and Systemic Evaluation
The workup of patients with retinal vasculitis necessitates suspicion for a wide variety of systemic infectious and inflammatory diseases (Table 10.1). In general, the only systemic infection that we routinely test for is syphilis, by sending a fluorescent treponemal antibody absorption (FTA-ABS) or equivalent assay. Retinal vasculitis can be the presenting finding an a number of autoimmune conditions (most of these are discussed further in Chapter 15), and thus this is one condition in which we order somewhat of a “shotgun” battery of immune related laboratory tests, including erythrocyte sedimentation rate, C-reactive protein level, rheumatoid factor, antinuclear antibody (ANA), antineutrophil cytoplasmic antibody, anti-DNA, anti-Smith, anticardiolipin antibodies, antiphospholipid antibodies, serum electrophoresis, serum cryoglobulins, complement levels, and anti-hepatitis B antibodies.
TREATMENT
Untreated, retinal vasculitis may lead to severe ocular complications such as macular edema, macular ischemia, peripheral vascular occlusion, retinal neovascularization, optic nerve atrophy, neovascular glaucoma, and retinal detachment. The goal of treatment for patients with retinal vasculitis is the suppression of intraocular inflammation and the prevention of visual loss and its long-term ocular complications. Immediate control of intraocular inflammation and retinal vasculitis is often achieved with periocular or intravitreal triamcinolone acetonide injections. Bilateral disease is often managed with systemic corticosteroids. Candidates for corticosteroid-sparing immunosuppression include patients with severe associated comorbidities or intolerance to corticosteroid therapy. Early and aggressive therapy can be critical for the prevention of irreversible visual loss. First-line immunomodulation therapy commonly involves methotrexate, azathioprine, cyclosporine, and mycophenolate mofetil. In cases of severe vision-threatening retinal vasculitis with associated systemic vasculitis, an alkylating agent such as cyclophosphamide or chlorambucil can be used. Addition of a systemic nonsteroidal anti-inflammatory drug can be used to reduce the rate of recurrence and treat the inflammation-associated CME. More recently, biologic drugs such as infliximab and daclizumab have offered additional options for patients of ocular and systemic autoimmune disorders with retinal vasculitis.
VESSEL INVOLVEMENT IN RETINAL VASCULITIS
Arteritis
- Systemic lupus erythematosus (SLE)
- Polyarteritis nodosa (PAN)
- Herpes simplex virus
- Herpes zoster virus
- Syphilis
- Churg-Strauss syndrome
Phlebitis
- Sarcoidosis
- Eales disease
- Toxoplasmosis
- HIV
- Birdshot retinochoroidopathy
- Paraviral syndrome
Both Arteries and Veins
- Wegener granulomatosis
- Multiple sclerosis
- Behçet disease
ANTIPHOSPHOLIPID SYNDROME
Overview
Antiphospholipid antibodies are a family of immunoglobulins (IgG, IgM, IgA, or mixtures) that recognize protein phospholipid complexes in laboratory tests. This syndrome consists of two different types of antibodies, lupus anticoagulant (LA) and the anticardiolipin antibody (ACA). ACAs are five times more common than LA. LA was first discovered in patients with SLE and thus the name. Both LA and ACA may be associated with arterial and venous thromboses, spontaneous abortion, and thrombocytopenia. Both antiphospholipid syndromes can be seen in association with SLE and other connective tissue disorders, malignancies, and infectious diseases. In a majority of cases, LA and ACA are not associated with any systemic disease and are found in healthy adult patients. While both arterial and venous occlusions can occur, venous occlusions are much more likely. It is often detected in females with recurrent miscarriages during pregnancy. Roughly 2% of the general population has antiphospholipid antibodies with a higher frequency in the elderly, while the prevalence is 30% to 40% in the patient with SLE.
Clinical Findings
Systemic Findings
- History of deep vein thrombosis, pulmonary embolism, transient ischemic attack, and myocardial infarction
- Recurrent miscarriages
- History of hematologic disorders
- Pulmonary hypertension
Ocular Findings
- Branch and central retinal artery and vein occlusions in otherwise healthy young adults
- Retinal hemorrhage
- Cotton wool spots
- Choroidal vascular occlusion
- Optic disc edema
Late Ocular Findings
- Peripheral and optic disc neovascularization due to a large area of peripheral retina ischemia
- Macular and retinal ischemia with sclerotic and occluded retina vessels
Differential Diagnosis
- Hypercoagulable states (protein S, protein C, antithrombin III, and heparin cofactor II deficiencies and factor V Leiden)
- Hyperviscosity syndrome (polycythemia, lymphoma, leukemia, thrombocythemia, paraproteinemia, and nephrotic syndrome)
- Sickle cell retinopathy
- Diabetic retinopathy
- Hyperhomocysteinemia
- Oral contraceptives
- Pregnancy
- Sarcoidosis
Diagnostic Testing
- Serum assay for LA and ACA IgG or IgM antibodies 12 weeks apart
- Serologic testing for autoimmune connective tissue diseases and systemic vasculitis
Treatment
- Aspirin therapy
- Anticoagulation therapy
- Panretinal photocoagulation for peripheral and optic disc neovascularization
- Intravitreal triamcinolone for retinal and macular edema
- Intravitreal bevacizumab for peripheral and optic disc neovascularization
EALES DISEASE
This disease is named after Henry Eales who, in 1880, described five young men with recurring vitreal and retinal hemorrhages that were associated with constipation and epistaxis. The definition of the disease has changed over the years. Most commonly, Eales disease is an idiopathic occlusive vasculopathy of the peripheral retina with areas of nonperfusion, neovascularization, and vitreous hemorrhage. In essence, the disease is a diagnosis of exclusion. The disease is most common in the Middle East and in India. Due to its high prevalence in these regions of the world, an association to tuberculosis infection has been speculated. In some clinical studies, a strong association with a positive purified protein derivative (PPD) and Mantoux test have further suggested the link between tuberculosis and Eales disease. The age of onset ranges from 17 to 44 years, with the mean age being 30 years. Patients often present with unilateral disease with the other eye involved in two thirds of cases. The average interval between the initial presentation and the involvement of the second eye is roughly 2.9 years. It is estimated to affect 1% of adult males in India younger than 40 years of age. Arteries are not involved and vitritis is minimal or absent. The periphlebitis is usually extensive but is irregular and noncontiguous. Overall, more than 50% of patients maintain a visual acuity of 20/50 or better.
Clinical Findings
- Peripheral retinovascular occlusion
- Extraretinal neovascularization is seen in 85% of patients
- Vitreous hemorrhage
- Venous tortuosity and vascular remodeling are present late
Four stages
- Ladder-like capillaries and mild periphlebitis associated with retinal edema
- Large vessel involvement with peripheral nonperfusion
- Neovascularization, retinal hemorrhage, and vitreous hemorrhage
- Scar tissue formation and traction
Differential Diagnosis
- Sarcoidosis
- SLE
- Temporal arteritis
- Multiple sclerosis
- Familial exudative vitreoretinopathy
- Behçet disease
Diagnostic Testing
- Complete blood count
- PPD and chest radiograph to rule out sarcoidosis and tuberculosis
- Autoimmune vasculitis workup
Fluorescein Angiography
- Vascular dilatation with staining of the occlusion (Fig. 10.3)
- Area of peripheral nonperfusion
- Microaneurysms
- Vascular remodeling and neovascularization with extensive leakage
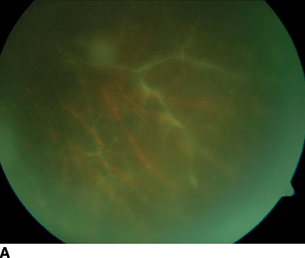
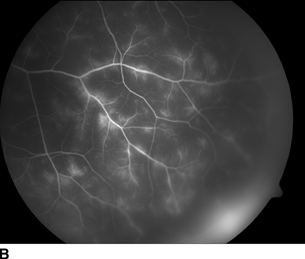
Figure 10.3 A: Fundus photo of retinal vasculitis demonstrates sclerosis of the retinal vessels with poor or no perfusion through the inflamed vessels. B: Fluorescein angiogram demonstrates extensive staining and late leakage from the inflamed vessels.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


