18
Masquerade Syndrome
John J. Huang and Anthony J. Correnti
INTRODUCTION
The masquerade syndromes classically refer to a group of neoplastic and paraneoplastic processes that cause intraocular infiltration of cells, mediator, and antibodies resembling immune-mediated uveitis. These cases are often initially misdiagnosed as “chronic idiopathic uveitis” upon referral. Masquerade syndrome can also involve disease entities such as retinitis pigmentosa, ocular ischemic syndrome, and chronic peripheral retinal detachment. Primary intraocular lymphoma (PIOL) is one of the most common, yet difficult to diagnose neoplastic masquerade syndromes. Masquerade syndromes should be considered in any patient over the age of 40 with persistent idiopathic uveitis that is nonresponsive to corticosteroids (Tables 18.1 and 18.2). Early recognition will allow for prompt treatment of the ocular disease, and potentially life-saving systemic treatment in cases of underlying malignancy.
Table 18.1 Masquerade syndromes

aConsider in childhood.
Table 18.2 Frequency of masquerade syndromes upon referral
Masquerade syndromes account for almost 5% of uveitis referrals to tertiary uveitis referral centers, with approximately half of the cases found to have an underlying malignancy. Overall age, sex, and racial distribution are dependent upon the underlying disease. The masquerade syndromes have a variable clinical presentation with vitreous and/or anterior chamber cells being the typical unifying feature. In malignant processes, other ocular features may include isolated chorioretinal lesions, serous retinal detachment, and iris infiltration. In nonmalignant diseases, findings such as chorioretinal lesions with surrounding cellular infiltrate and pseudo-retinal vasculitis, may be present.
DIAGNOSIS
Guidelines for workup of idiopathic ocular inflammation are dependent on number of variables, including preexisting systemic disease, patient age, and clinical examination. This is often accomplished with the cooperation of the ophthalmologist and the primary care physician. Further ancillary testing, including fluorescein angiography, ultrasonography, optical coherence tomography, and electro-retinogram (ERG), should be ordered on the basis of patient’s suspected diagnosis. Neuroimaging and lumbar puncture are required for the diagnosis of PIOL. Intraocular fluid analysis, including vitreous and anterior chamber aspirate, should be considered in cases of suspected malignancy.
TREATMENT
Treatment of the masquerade syndromes varies based upon the underlying etiology, and requires a multidisciplinary approach. Empiric systemic corticosteroids can temporarily resolve pseudoinflammation, but may mask the underlying diagnosis. In malignant disease, collaboration amongst the ophthalmologist, oncologist, and internist is crucial, as timely intervention can improve morbidity outcomes.
ADENOMA/ADENOCARCINOMA OF THE RETINAL PIGMENT EPITHELIUM
Adenoma/adenocarcinoma of the retinal pigment epithelium (RPE) is a rare tumor that can be aggressive clinically. They are most often diagnosed pathologically upon enucleation for suspected choroidal melanomas, rather than at the time of clinical examination. Patients are asymptomatic or present with decrease vision. Most lesions are discovered incidentally or referred for presumed choroidal melanoma. Important distinguishing features include dark black color, dome shaped, vitreous cell, exudative retinal detachment, and retinal feeder vessels.
Clinical Signs
- Jet-black solitary lesions (rarely amelanotic)
- Abruptly elevated
- Dome shaped
- Unilateral
- Retinal feeder vessels (sometimes)
- Vitreous cell
- Vitreous hemorrhage
- Exudative retinal detachment
Differential Diagnosis
- Choroidal melanoma
- Choroidal nevus
- Melanocytoma
- Combined hamartoma of the retina and RPE
- Congenital hypertrophy of the RPE
- Reactive hyperplasia of the RPE
- Choroidal detachment
Diagnostic Testing
Fluorescein Angiogram
- Early hypofluorescence with minimally late hyperfluorescence—Presence of retinal feeder vessels
Ultrasonography
- A-scan: medium to high internal reflectivity
- B-scan: dome shaped, “stuck on” appearance with acoustic solidarity
Fine-Needle Aspiration Biopsy
- Useful to differentiate from choroidal melanoma
Treatment
- Observation with periodic examination (in asymptomatic lesions)
- Consider local resection in progressive, symptomatic lesions
CANCER-ASSOCIATED RETINOPATHY
First described in 1976 by Sawyer in three patients with rapid vision loss, visual phenomena, ring scotomata, and nyctalopia; later they were diagnosed with lung carcinoma. Cancer-associated retinopathy (CAR) is a rare autoantibody-mediated paraneoplastic disease that involves tumor antigen cross-reactivity with retinal photoreceptor recoverin protein. Histologic studies demonstrated degeneration of the outer nuclear retinal layer and disintegration of photoreceptors and the presence of macrophages. CAR is typically seen in older adults without a predilection for sex. Clinical findings are often absent or subtle, and patients often present without a known diagnosis of systemic malignancy. Patients present with a variety of symptoms such as visual loss, constriction of visual field, nyctalopia, flickering light sensation, and photophobia. In general, the ocular symptoms antedate the diagnosis of systemic disease by 5 months. The typical patients progress to blindness in 6 to 18 months. No treatment has been consistently effective for the treatment of this condition. CAR should be suspected in the setting of unexplained bilateral vision loss and constriction of the visual field in elderly population.
Clinical Signs
- Nonspecific pigmentary mottling in the macula
- Vascular attenuation
- Optic disc pallor
- Vitreous cells (rare)
Differential Diagnosis
- Recoverin-associated retinopathy (no evidence of systemic malignancy)
- Melanoma-associated retinopathy (MAR)
- Acute zonal occult outer retinopathy
- Cone dystrophy
- Retinitis pigmentosa
- Arteritic and nonarteritic ischemic optic neuropathy
- Toxic optic neuropathy
- Hereditary optic neuropathy
- Nutritional optic neuropathy
- Retrobulbar optic neuritis
Diagnostic Testing
Visual Field Testing
- Peripheral field constriction (Goldmann)
- Paracentral scotoma
Electroretinogram (Full-Field)
- Usually markedly abnormal photopic (cone) waveforms
- Also may have decreased scotopic (rod) responses or combination
- Both A and B waves are flat
Fluorescein Angiography
- Helpful in ruling out other causes of vision loss; no specific pattern in CAR
Optical Coherence Tomography
- Thinning of inner retinal layers
Lab Studies
- Western blot and immunofluorescent antibody assay for antiretinal antibodies (essential for diagnosis).
- If there is no known malignancy, perform complete history and physical exam. Consider chest X-ray, CT scan of abdomen/pelvis/chest, mammography (woman), and total-body positron emission tomography.
Treatment
- High-dose corticosteroids may provide short-term benefit with improvement of symptoms.
- Immunosuppressive therapy.
- Plasmapheresis.
- Intravenous immunoglobulin therapy.
- Treatment of underlying systemic malignancy may not improve visual outcome.
CHOROIDAL HEMANGIOMA (CIRCUMSCRIBED TYPE)
Choroidal hemangioma is benign vascular tumor that has been described in two clinical forms: circumscribed and diffuse. Diffuse choroidal hemangioma is classically associated with Sturge-Weber syndrome, while circumscribed lesions generally have no systemic association. First described by Leber in 1868 of a patient enucleated for pain. In most clinical series, 50% of choroidal hemangioma is described as circumscribed and 50% as diffuse form associated with Sturge-Weber. Choroidal hemangioma are likely congenital in origin, and are most often seen in Caucasians with no gender predilection. The symptomatic patients commonly present in the range 20 to 50 years of age. The choroidal hemangioma and associated subretinal fluid produce symptoms of metamorphopsia, induced hyperopia, blurred vision, and visual field defects. The visual loss in patients with choroidal hemangioma is related to the retinal degeneration (retinal edema, cystic degeneration, loss of photoreceptors, gliosis, and RPE migration) seen even in small tumors. Most important factor for visual prognosis is location of the hemangioma.
Clinical Signs
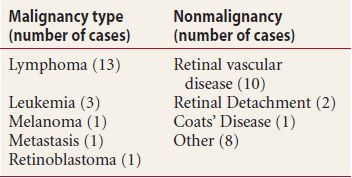
Elevated, orange-red, flesh-colored, mottled orange, or grayish lesion (Fig. 18.1)
- Usually located posterior to the equator, near optic disc
- Unilateral and solitary
- Poorly defined margins
- Amelanotic and transilluminate
- Overlying serous detachment (may involve macula)
- May have brown pigment on tumor surface
- Cystoid macular edema
- Retinal degeneration overlying the tumor
- Choroidal neovascular membrane (rare)
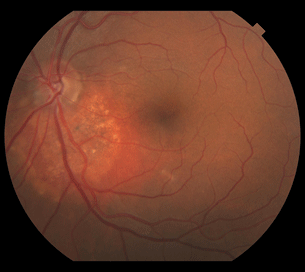
Figure 18.1 Patient with circumscribed peripapillary choroidal hemangioma. The lesion is orange in appearance with associated pigmentary changes of the RPE and subretinal fluid.
Differential Diagnosis
- Amelanotic choroidal melanoma
- Choroidal metastasis
- Choroidal neovascularization (CNV) with disciform scarring
- Choroidal osteoma
- Posterior scleritis
- Central serous chorioretinopathy
- Retinoblastoma
Diagnostic Testing
Fluorescein Angiogram
- Early coarse pattern of spotty hyperfluorescence during choroidal filling
- Late confluence of hyperfluorescence with blurred margins (Fig. 18.2)
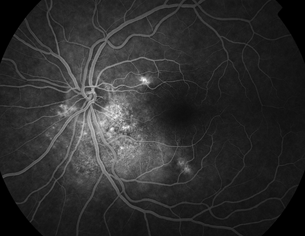
Figure 18.2 Fluorescein angiogram demonstrates increased hyperfluorescence due to numerous pinpoint areas of leakage associated with the choroidal hemangioma.
Ultrasonography
- Elevated subretinal mass with acoustic solidarity and high internal reflectivity (characteristic finding)
Indocyanine Green Angiography
- Early, well-defined, area of intense, homogenous hyperfluorescence
- Intrinsic tumor vessels
- Late “dye washout” with loss of tumor hyperfluorescence
Systemic Workup
- In diffuse cases, workup for Sturge-Weber and glaucoma
Others
- Color Doppler Imaging (high Doppler shifts)
- Confocal laser scanning fluorescence topography
Treatment
- Observation in non–vision threatening lesions with no macular involvement
- Photodynamic therapy (preferred treatment in cases with serous detachment threatening/involving the fovea)
- Laser photocoagulation is highly effective, but 40% of lesion reaccumulate the subretinal fluid and will need retreatment
- External bean irradiation
- Brachytherapy
CHOROIDAL MELANOMA
Choroidal melanoma is the most common primary intraocular malignancy among adults with incidence of six cases per million per year. In the United States, the incidence of choroidal melanoma is 1/8 of cutaneous melanoma. The disease is often diagnosed in the sixth decade of life with the incidence increasing with age. Factors associated with increased mortality include tumor size, location, transscleral/optic nerve extension, amelanotic lesions, growth through Bruch membrane, increased patient age, and histopathologic type. Most symptomatic melanomas present because of decreased vision, flashes, floaters, or visual field loss due to tumor growth near the optic nerve or fovea, leading to exudative retinal detachment. Pain is an extremely uncommon unless there is associated glaucoma, intraocular inflammation, or extraocular extension.
Clinical Signs
- Elevated, sub-RPE mass (Fig. 18.3)
- Variable pigmentation, amelanotic (yellow-white) to darkly pigmented in color
- Overlying RPE changes: orange pigment (highly characteristic), drusen, atrophy
- Well-circumscribed
- May be multilobed or assume mushroom configuration
- Overlying serous or exudative detachment
- Minimally elevated dark choroidal lesion without defined border (diffuse melanoma)
- Vitreous cells
- Vitreous or subretinal hemorrhage
- Dilated episcleral vessels (sentinel vessels) in anterior lesions
- Conjunctival hyperpigmentation (transcleral extension)
- Predisposing ocular diseases:
- Choroidal nevus
- Congenital ocular melanocytosis
- Dysplastic nevus syndrome
- Xeroderma pigmentosum
- Choroidal nevus
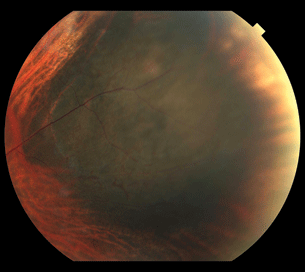
Figure 18.3 Large peripheral choroidal melanoma with associated subretinal fluid and intrinsic vessels within the tumor.
Differential Diagnosis
- Melanotic lesions:
- Choroidal nevus (Fig. 18.4)
- Melanocytoma
- Adenoma/Adenocarcinoma of RPE
- Combined hamartoma of the retina and RPE
- Congenital hypertrophy of the RPE
- Reactive hyperplasia of the RPE
- Choroidal detachment
- Choroidal nevus (Fig. 18.4)
- Amelanotic lesions:
- Choroidal hemangioma
- Amelanotic choroidal nevus
- Choroidal metastasis
- CNV with subretinal scarring
- Choroidal osteoma
- Lymphoma
- Posterior scleritis
- Choroidal hemangioma
Figure 18.4 Small flat choroidal nevus in the posterior pole. There is no elevation, no evidence of subretinal fluid or superficial lipofuscin.
Diagnostic Testing
Transillumination
- Shine light through the pupil, with blockage of transillumination by the melanoma
Fluorescein Angiogram
- Variable; may be normal angiogram
- Early patchy hypofluorescence or hyperfluorescence with late staining
- May show “double circulation” pattern of combined choroidal and retinal filling (highly characteristic)
Ultrasonography
- A-scan: low-medium internal reflectivity; oscillations of internal spike
- B-scan: elevated subretinal mass with low-medium internal reflectivity, excavation, and/or shadowing of underlying tissues
- Useful for documenting tumor thickness (usually >3 mm) and basal diameter (>6 mm)
Metastatic Workup
- Complete physical exam
- Liver function tests (consider liver scan if positive)
- Chest X-ray
Others
- CT/MRI
- PET scan
- Indocyanine green angiography
- Fine needle biopsy
Treatment
- Consultation with oncologist/ocular oncologist
- Laser photocoagulation
- Radiotherapy
- Transpupillary thermotherapy
- Brachytherapy
- Local resection
- Enucleation
METASTATIC CANCER
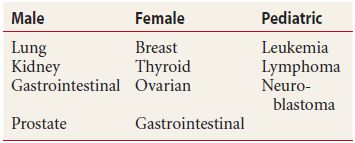
Metastatic cancer is considered the most common intraocular malignancy. The choroid due to the high level of blood flow is the most common site of metastasis, although retinal, optic nerve, and vitreous involvement can occur. The embolic tumor cells will require the local production of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor for tumor growth. Clinical appearance of the lesion is dependent on the type of primary cancer, site of intraocular involvement, and the extent of secondary changes. Overall, breast and lung cancer are the most common primary cancer. A primary site is known in approximately two-thirds of cases at the time of diagnosis, with 17% of cases remaining unknown after thorough systemic evaluation (Table 18.3). The period of survival ranges from 2 weeks to 5 years with an average of 9.5 months. The duration of survival reflects the nature of the primary cancer and the extent of systemic metastasis.
Table 18.3 Current primary cancers of choroidal metastasis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


