15
Systemic Diseases Associated
with Ocular Inflammation
Deanne Nakamoto and Paul A. Gaudio
This chapter aims to serve as a succinct reference for ophthalmologists treating patients with systemic collagen vascular diseases (CVDs) and systemic infectious diseases.
COLLAGEN VASCULAR DISEASES
In treating all collagen vascular diseases, keep in mind
- The most common manifestation of almost all CVDs is dry eyes.
- Ocular inflammation indicates that a CVD is severe and requires increased therapy, regardless of how the rest of the patient’s body appears to be doing. It is important to communicate this with the patient’s other care providers.
- In patients with CVD, ocular inflammation is generally more difficult to treat than inflammation in other organ systems, and requires higher doses of anti-inflammatory medications. Patients are well served when ophthalmologists are comfortable adjusting medication doses to optimize therapy, or at the very least when ophthalmologists have very close working relationships with rheumatologists.
- The immunosuppressive regimens used to treat CVDs commonly result in opportunistic ocular infectious, which can present in atypical forms and be quite severe. Herpes and toxoplasmosis are probably the most common pathogens in this setting.
Rheumatoid Arthritis
Rheumatoid arthritis (RA) is mainly an inflammatory joint disease, but extra-articular involvement is not uncommon, and nearly all organ systems can be affected. Women are affected three times more than men, mostly aged 40 to 60. There is at least some genetic predisposition conferred in a handful of specific HLA-DQ and HLA-DR alleles. Morning stiffness is the hallmark and probably is the most widely noted presenting symptom.
Presentation
There are specific diagnostic criteria for RA. Patients need four of the seven findings given below:
- Morning stiffness for greater than 1 hour.
- Symmetric arthritis.
- Arthritis of the hands. Patients will often have boggy, warm knuckles. Advanced disease is notable for the radial deviation of the wrist and ulnar deviation of the digits, swan neck digits, and boutonniere’s deformity (Fig. 15.1).
- Arthritis in three or more joints. Usually the spine (except cervical) is spared.
- Rheumatoid nodules.
- Elevated rheumatoid factor (RF). RF is an antibody against the Fc portion of IgG, found in 5% of normal individuals and two thirds of RA patients.
- Joint erosions detectable on hand or wrist X-ray.
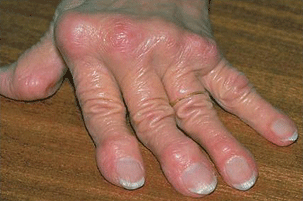
Figure 15.1 Elderly patient with RA with a chronic history of inflammation of the hands. There is severe deviation of the joints with swan neck digits.
Ophthalmic manifestations are the following:
- Dry eye is the most common eye finding.
- Scleritis. Rheumatoid patients are at special risk for necrotizing scleritis (Fig. 15.2) and scleromalacia perforans.
- Episcleritis.
- Keratitis. Rheumatoid keratitis can present as a sterile, avascular melt, often with very little stromal infiltrate, which can lead to perforation. PUK and necrotizing scleritis (Fig. 15.3) in this setting indicate potentially fatal systemic disease requiring powerful immunosuppressive therapy.
- Neither primary uveal tract inflammation nor retinal vasculitis is generally considered part of the RA complex, though some patients (about 30%) with scleritis will have uveitis secondarily.
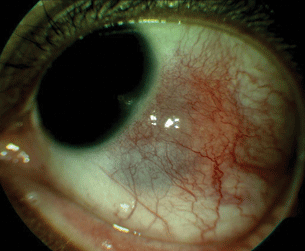
Figure 15.2 RA-associated nodular scleritis with associated scleral thinning.
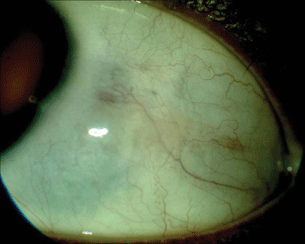
Figure 15.3 After aggressive treatment, the nodular scleritis has resolved with residual area of scleral thinning.
Laboratory Findings
- The main laboratory findings are elevation in RF and cyclic citrullinated protein antibodies.
- Elevated ESR and normocytic, normochromic anemia are also common.
Treatment
- NSAIDS, for symptomatic joint relief.
- Prednisone, often under 10 mg for long periods.
- Hydroxychloroquine (Plaquenil), typically 100 to 400 mg per day (it should not exceed 6.5 mg/kg/day).
- Sulfasalazine.
- Methotrexate, 7.5 to 25 mg per week.
- Cyclophosphamide in severe cases.
- More recently, a number of biologic agents have been developed with greater efficacy and fewer side effects than traditional therapies. These include the tumor necrosis factor alpha (TNF-α) blockers etanercept, infliximab, and adalibumab, along with other compounds that target various immune-signaling mechanisms.
Monitoring Disease Activity
- Monitoring patients with RA involves mostly following patients’ joint findings and overall feeling of well-being.
- The American College of Rheumatology (ACR) has scoring systems referred to as ACR 20 and ACR 50 in an effort to quantify disease activity for research purposes.
- RF is not a reliable long-term parameter of disease progression.
- Death from RA is usually the result of vaso-occlusive diseases affecting the heart and brain.
Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is a multisystem disease in which individuals produce self-directed antibodies. These antibodies cause widespread tissue and cell destruction via the formation of immune complexes and the misdirection of the inflammatory response. Women are four times more commonly affected than men, mostly in the childbearing years. The disease is more common in black, Hispanic, and Asian women than in whites. SLE can affect the entire body, so when evaluating a patient we find it very helpful to clarify which organs are involved and to what extent.
Presentation
- Most patients will note skin, kidney, or joint involvement, but the range of symptoms and physical findings is very broad.
- Four of the following 11 criteria are considered diagnostic of SLE:
- Malar rash on the bridge of the nose and cheeks.
- Discoid skin lesions, roughly dime sized, red, slightly indurated.
- Photosensitivity when the skin is exposed to UV light.
- Pleuritis or pericarditis.
- Arthritis.
- Nasal or pharyngeal ulcers.
- Seizures or psychosis.
- Hematologic abnormalities: anemia, thrombocytopenia, and leucopenia.
- Renal abnormalities: proteinuria and nephritis.
- ANA elevation. Titers of 1:160 or higher are significant.
- Other serologic abnormalities, for example, autoantibodies such as anti–double-stranded DNA and anti-Smith.
- Malar rash on the bridge of the nose and cheeks.
- Ocular involvement
- Dry eye (Fig. 15.4).
- Scleritis.
- Episcleritis.
- Keratitis.
- Ischemic retinopathy and choroidopathy including retinal hemorrhages, cotton-wool spots, and artery occlusions.
- Purely uveal tract inflammation is an uncommon eye finding, although elevated ANA is observed in some uveitis patients who do not have SLE.
- Dry eye (Fig. 15.4).
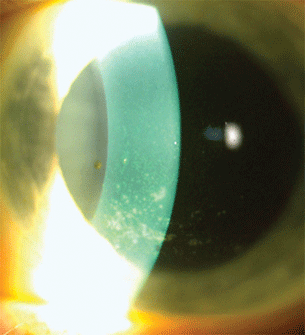
Figure 15.4 SLE patient with secondary SS with severe dry eyes and superficial punctate keratopathy.
Laboratory Findings
- ANA elevation (1:160 or higher is significant). More than 95% of SLE patients have this.
- Anti–double-stranded DNA; anti-Sm.
- False positive VDRL.
- Unfortunately, none of the antibody tests have been shown to uniformly indicate disease status. Disease activity is assessed according to patients’ symptoms, their physical examination, and organ-specific laboratory findings.
Treatment
- Hydroxychloroquine—This drug may cause foveal toxicity (Bull’s eye maculopathy) at doses over 6.5 mg/kg/day, and its use is an indication for the periodic evaluation of retinal function (we check a 10 degree automated visual field with a red target).
- Prednisone for immediate control, and many patients take low doses for prolonged periods.
- Methotrexate.
- Azathioprine.
- Mycophenolate mofetil.
- Cyclophosphamide.
- Rituximab, and at times also infliximab.
- In patients treated with immunosuppressive therapy for SLE, we maintain a high index of suspicion for herpetic keratitis and herpetic and toxoplasma retinitis occurring as secondary infections.
- Plasmapheresis is an intervention reserved for severe acute SLE flares. It involves removal of the plasma, the portion of blood containing antibodies.
Primary Antiphospholipid Syndrome (PAPS)
The term “antiphospholipid antibodies” refers to antibodies against phospholipid-protein complexes on cell membranes. The presence of these antibodies is considered pathologic, and predisposes a person to excessive thrombosis. Many patients who have these antibodies have SLE or other autoimmune diseases. In the absence of a connective tissue disease, the term “primary antiphospholipid syndrome” (PAPS) is used. While antiphospholipid antibodies are found in roughly 1–5% of the population (and nearly 50% of SLE patients), the full syndrome termed PAPS is rare.
Presentation
PAPS is marked by the presence of one of the following three clinical findings plus the presence of antiphospholipid antibodies on laboratory testing. Clinical findings include
- Arterial or venous thrombosis, or small vessel vaso-occlusive disease
- Pregnancy morbidity (fetal death, premature birth due to preeclampsia or placental insufficiency, or three first trimester losses)
- Ophthalmic manifestations
- Retinal vascular occlusions (Fig. 15.5)
- Any form of ocular inflammation
- Retinal vascular occlusions (Fig. 15.5)
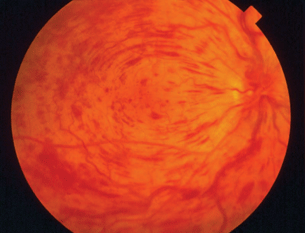
Figure 15.5 Patient with PAPS with central retinal vein occlusion
Laboratory Findings
- Three types of clinically defined antiphospholipid antibodies exist:
- Anti-cardiolipin IgM or IgG detected by ELISA assay on two separate occasions 12 weeks apart.
- Anti-β2 glycoprotein-1 IgM or IgG, also detectable by ELISA.
- The “lupus anticoagulant,” this being any of a number of autoantibodies detected by liquid-phase coagulation assays in which the clotting time is prolonged (e.g., Russell venom viper test).
- Roughly 50% of patients with SLE will have one of the antiphospholipid antibodies, but the term “primary antiphospholipid syndrome” is used when these antibodies are found in patients who have no other evidence of SLE.
- Thrombocytopenia is, paradoxically, often observed in patients with PAPS.
- Anti-cardiolipin IgM or IgG detected by ELISA assay on two separate occasions 12 weeks apart.
Treatment
- Corticosteroids and immunosuppression as other forms of uveitis.
- Anticoagulation to prevent further vaso-occlusive events.
Sarcoidosis
Sarcoidosis is a relatively common immune-mediated disease in which T-cells and macrophages proliferate and collect in virtually every organ system. The immune aggregates eventually organize into the characteristic non-caseating granulomas and mechanically disrupt the structure of vital organs. Sarcoidosis most frequently presents in the lung and the eye. It affects all races and both genders, but in the United States, the majority of sarcoidosis patients are black, with a prevalence of 40 per 100,000 compared to 5 per 100,000 among whites, roughly an 8:1 ratio. Interestingly, this trend does not hold elsewhere in the world. Globally, the disease is most prevalent in Northern European countries, and the highest annual incidence of 60 per 100,000 is found in Sweden and Iceland. Most patients present between 20 and 40 years of age.
The systems most commonly affected by sarcoid are the lungs, skin, and eyes. The clinical course varies and ranges from asymptomatic disease that resolves spontaneously to a debilitating chronic condition.
Presentation
- Systemic manifestations
- Shortness of breath. Cough, dyspnea, and chest pain; especially on exertion. Almost all patients will have lung symptoms and/or a positive chest X-ray at some point (Fig. 15.6).
- Lymphadenopathy.
- Erythema nodosum (Fig. 15.7).
- Blood abnormalities due to bone marrow involvement.
- Constitutional symptoms: fever and fatigue.
- Joint pains.
- Lofgren syndrome is the constellation of erythema nodosum, febrile arthropathy, bilateral hilar lymphadenopathy, and iritis.
- Heerfordt syndrome (uveoparotid fever) is the name given to sarcoidosis with uveitis, parotitis, fever, and facial nerve palsy.
- Mikulicz syndrome is the term for sarcoidosis with lacrimal and parotid swelling and keratoconjuntivita sicca.
- Shortness of breath. Cough, dyspnea, and chest pain; especially on exertion. Almost all patients will have lung symptoms and/or a positive chest X-ray at some point (Fig. 15.6).
- Ophthalmic manifestations (40% of patients will have eye findings).
- Anterior granulomatous uveitis. Eyes will often have mutton fat keratic precipitates (KP) and iris nodules.
- Intermediate uveitis.
- Posterior uveitis. Candlewax drippings, cystoid macular edema, and periphlebitis are the most common findings (Fig. 15.8). Some patients will develop yellow-orange round retinal spots scattered diffusely in the periphery, roughly 300 µm in size, often rather subtle against the orangish glow of the healthy choroid. A third fundus finding occasionally encountered is the presence of more discreet round chorioretinal nodules, roughly at the equator, that resolve to leave scars roughly 500 µm in size.
- External eye findings: Dacryoadenitis, millet seed lids, and conjunctival nodules.
- Anterior granulomatous uveitis. Eyes will often have mutton fat keratic precipitates (KP) and iris nodules.
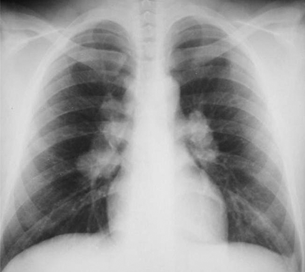
Figure 15.6 Patient with active pulmonary sarcoidosis with diffuse hilar adenopathy on chest radiography.
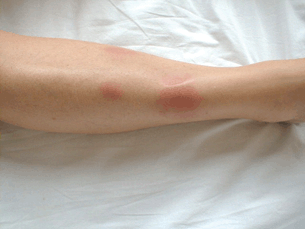
Figure 15.7 Patient with active sarcoidosis developed erythema nodosum lesion of the right lower leg.
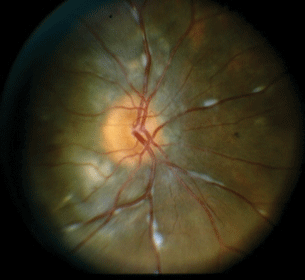
Figure 15.8 Sarcoidosis-associated posterior uveitis with optic nerve inflammation, periphlebitis, and underlying pigmentary changes with associated choroidal inflammation.
Laboratory Findings and Ancillary Testing
- Chest X-ray. Ninety percent will have an abnormal finding, mostly commonly hilar lymphadenopathy.
- Angiotensin-converting enzyme.
- Serum lysozyme.
- Gallium-67 scan of head and neck. The scan should be taken with the patient off steroids.
- Brochioalveolar lavage.
- Lesion biopsy. If there is a conjunctival nodule, biopsy it to look for non-caseating granulomas.
- Histopathologic findings. Non-caseating epithelioid cell granulomas with inclusion bodies. Granulomas are defined by the presence of epithelioid cells, which are polyhedral mononuclear histiocytes derived from monocytes (Fig. 15.9).
- RF and ANA can be falsely elevated.
- Other laboratory abnormalities according to organ involvement.
- Decreased WBC, hemoglobin with bone marrow involvement.
- Increased creatinine with kidney involvement.
- Decreased WBC, hemoglobin with bone marrow involvement.
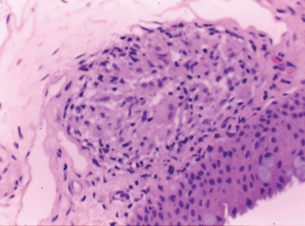
Figure 15.9 Histology of a sarcoid nodule with non-caseating granuloma.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


