Fig. 11.1
Eyelid – Low-power view of the eyelid showing normal skin appendage structures and tarsal plate containing Meibomian glands (hematoxylin and eosin stain; original magnification 20×)
11.2 Normal Histology
The outermost layer is thin skin, and the epidermis consists of a stratified squamous epithelium with some papillae. The epidermis is composed of two cell types: the keratinocytes and the dendritic cells. The keratinocytes are arranged in four layers; the deepest is the basal cell layer. The squamous cell layer (stratum spinosum) and granular layer are intermediate, and the nonnucleated keratinized layer is the most superficial. The term “stratum malpighii” is most often used to describe the lower three nucleated layers.
The basal keratinocytes form a single row of cells resting on a basement membrane, which is intimately attached to the dermis. The cells are columnar with their long axes arranged perpendicular to the skin surface. They are connected to each other by intercellular bridges and may normally contain variable amounts of melanin pigment derived, in part, from adjacent dendritic melanocytes. The squamous cell layer is composed of a mosaic of polygonal keratinocytes that flatten superficially where they run parallel to the surface. The granular layer forms a row of elongated flat cells containing coarse basophilic keratohyalin granules. The external horny layer consists of flat keratinized cells devoid of nuclei.
There are three types of dendritic cells present in the epidermis: (1) clear cell melanocytes, (2) Langerhans cells, and (3) undetermined dendritic cells.
The dermis is loose and delicate. It contains hair follicles as well as sebaceous and sweat glands. The dermis is composed of a papillary layer and a reticular layer. The papillary dermis forms numerous cone-shaped dermal papillae, which extend upward into the epidermis. The ridges of epidermis separating the papillae are called rete ridges. The reticular dermis contains thick bundles of collagen and variable amounts of elastic and reticulin fibers as well as ground substance containing mucopolysaccharides. Small nerve fibers, blood vessels, and lymphatics are also found in the reticular dermis.
The subcutaneous layer of the eyelids contains a small amount of adipose tissue. It is loosely adhered to the underlying orbicularis muscle. For this reason, swelling of the lids (commonly caused by edema, hemorrhage, or acute inflammation in this layer) is easily visible clinically.
The inner layer is a mucous membrane, the palpebral conjunctiva. The lining epithelium is a low stratified columnar type with goblet cells scattered among the epithelial cells. The stratified squamous epithelium of the skin continues over the margin of the eyelid and is transformed into the stratified columnar type. The thin lamina propria of the palpebral conjunctiva contains connective tissue with elastic and collagenous fibers. Beneath the lamina propria is a plate of dense, collagenous connective tissue, the tarsal, which contains large, specialized sebaceous glands, the tarsal (meibomian) glands. They secrete sebum, which is extruded into the meibomian ducts, the orifices of which open on the lid margin just behind the gray line. This sebaceous material contributes to the lipid component of the tear film.
The free end of the eyelid contains eyelashes, which arise from large, long hair follicles. Associated with the eyelashes are small sebaceous glands (Zeis glands). The apocrine glands of Moll, which empty their secretions into the follicles of the lashes, are located between the hair follicles and large sweat glands. The cells lining these glands show secretory apical granules and areas of decapitation.
Three sets of muscles are present in the eyelid: the extensive palpebral portion of the skeletal muscle, the orbicularis oculi, the skeletal ciliary muscle (of Riolan) in the region of the hair follicles of the eyelashes and tarsal glands, and the smooth, superior tarsal muscle (of Müller). The orbicularis oculi muscle forms an elliptic sheet of concentrically arranged, striated muscle fibers within the eyelids. In the upper lid, the tendons of the levator palpebrae superioris pass through it to insert into the skin and tarsus. A portion of the orbicularis muscle known as the muscle of Riolan consists of striated muscle fibers that lie adjacent to the tarsal plate near the lid margin. Portions of this muscle are located superficial to the meibomian glands, and the remainder (subtarsal portion) lies deep to them. The glands of Moll separate the Riolan muscle from the palpebral portion of the orbicularis. The smooth muscle of Müller in the upper lid lies adjacent to the orbital septum under the levator palpebrae superioris. It originates among the fibers of the levator in the upper lid and the inferior rectus in the lower. The fibers of the superior and inferior Müller’s muscle insert, in part, into the margins of the tarsal plates and also insert into collagenous tissue attached to the deep fibers of the orbicularis muscle. Alterations of these muscular structures may lead to ptosis, ectropion, or entropion.
A variety of epidermal appendages (glands and cilia) are present in the lids. The sebaceous glands are holocrine. Their acini possess no lumina and extrude their secretory products by decomposition of their cells. The meibomian glands are situated within the tarsal plates where they are arranged vertically and parallel to each other and are larger in the upper tarsus. The eccrine sweat glands are composed of three segments: a secretory portion, an intradermal duct, and an intraepidermal duct. The apocrine glands of Moll lie near the lid margin and empty their secretions into the follicles of the lashes. Accessory lacrimal glands with histological features identical to those seen in the main lacrimal gland are often found in the substantia propria of the conjunctiva. The glands of Krause are deeply located in the subconjunctival tissues at the fornices. All of these glands vary in number and are not seen in every section of the eyelid. The hair of the skin of the lid is scanty and fine.
11.3 Congenital and Developmental Abnormalities
Abnormal development and maturation of the ectoderm and mesoderm in the embryonic lid fold may result in several deformities of the eyelid and the palpebral aperture. These may be limited to the eyelid or associated to abnormalities in the globe, orbit, face, skull, and extremities. The severity of the anomaly can vary greatly, and usually the minor defects tend to be isolated to the eyelid region and are, in general, more common than the severe defects [2, 3].
11.3.1 Cryptophthalmia
Rare condition, unilateral or bilateral, it is the result of failure of the eyelid formation. It is characterized by a continuous layer of epidermis extending between the forehead and the cheek, covering a malformed eye in most cases. This condition most often occurs in the setting of multiple malformations involving the skull, midface, extremities, and urogenital tract [4].
11.3.2 Microblepharon
Congenital anomaly characterized by vertical shortage of the upper and/or lower eyelid skin causing nocturnal lagophthalmos, corneal exposure, and cosmetic deformity. This abnormality can be unilateral or bilateral [5].
11.3.3 Coloboma
Coloboma, from the Greek word koloboma, meaning defect, represents a focal disruption in the normal development of the eyelid. The severity of the defect can vary from a small notch in the eyelid margin to a near-total absence of the eyelid (Fig. 11.2). Eyelid colobomas may involve a single or all four lids. The maldevelopment might be related to an amniotic band impingement or faulty lid fusion [6]. As observed in other ocular colobomas, patients with eyelid coloboma have a mutation in the PAX2 gene [7]. Band-like adhesions may join the coloboma to the epibulbar conjunctiva. A continuous bridge of skin and conjunctival mucosa bridges the two unaffected edges of the eyelid. An abrupt loss of lashes, tarsus, and adnexal structures demarcates the borders of the coloboma.
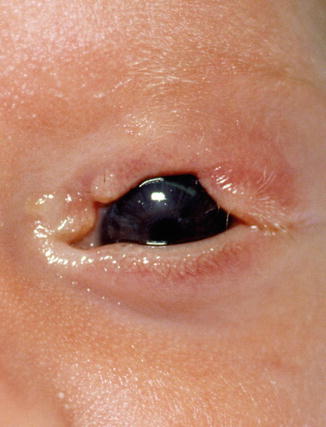
Fig. 11.2
Coloboma – eyelid coloboma in a small infant. The upper eyelid shows an area of absence of lashes and tarsus demarcating the edge of the defect (Clinical picture courtesy of Dr. George Bartley, Mayo Clinic)
11.3.4 Distichiasis
Rare disorder characterized by the presence of an extra row of eyelashes originating from the orifices of abnormally formed meibomian glands. This is the result of an anomalous development of the epithelial germ cells failing to differentiate completely to meibomian glands and instead becoming pilosebaceous units and associated with a mutation in FOXC2 gene [11, 12]. The Moll’s glands are usually hypertrophic and the tarsus plate absent or rudimentary. This abnormality is often associated with other malformations [3].
11.3.5 Phakomatous Choristoma
It represents an accessory rest of the surface ectoderm. It forms when cells from the lens placode do not accompany the normal migration into the optic cup and become entrapped into the connective tissue of the eyelid. It is also known as Zimmerman tumor, in honor of the author that first described this entity in 1971 [13]. Clinically, it presents consistently at birth as a palpable nodule in the inferonasal region of the lower eyelid [14, 15], adjacent to the lacrimal sac, occasionally causing nasolacrimal duct obstruction [16]. Histologically, it is characterized by cuboidal, lenslike epithelium surrounded by basal membrane material. These cells might proliferate within a collagenous stroma and also undergo cataractous changes. Ultrastructural studies and immunohistochemical profile show a strong positivity with vimentin and S-100 protein and have confirmed a lenticular origin [14, 16–18].
11.3.6 Xeroderma Pigmentosum
Xeroderma pigmentosum (XP) is a rare, autosomal recessive inherited disorder characterized by defective DNA repair to the damage caused by ultraviolet light, which manifests clinically as photosensitivity, actinic damage to the skin, propensity to early development of cutaneous and ocular neoplasms, and, in some patients, neurological degeneration [19]. Abnormalities affecting the eyelids, conjunctiva, and cornea (areas exposed to ultraviolet radiation) have been reported in 40 % of published cases [20, 21]. The disease affects both sexes equally. It is rare in North America and Europe but is more common in areas with increased consanguinity such as Japan, the Middle East, and India [22, 23].
A defective gene locus has been identified in all seven complementation groups (A–G) and the one variant form of XP [20, 24]. In the complementation group A (MIM +276700), the classical form of XP [mutations located on both chromosome 1 and chromosome 9 (9q22)] have been identified.
The genetic defect causes mutation of nucleotide excision repair (NER) enzymes, leading to a reduction in or elimination of NER [25]. Damage to an epidermal cell’s DNA occurs during exposure to ultraviolet (UV) light. If a tumor suppressor gene such as p53 or proto-oncogene is affected, a neoplasm might develop. Patients with XP are at a high risk for developing a variety of cutaneous cancers, such as basal cell carcinoma, squamous cell carcinoma, as well as malignant melanoma [26, 27]. The eyelids are frequently affected. Most patients die before the age of 21 years from metastatic squamous cell carcinoma or melanoma [28, 29].
Eyelid cutaneous changes reflect the local actinic damage (Fig. 11.3), including erythema, hyperpigmentation, atrophy, scarring, and development of neoplasms such as basal cell carcinoma, squamous cell carcinoma, and melanoma in a very young age [20]. These neoplasms have been reported to be multiple and bilateral when affecting a same patient [21].
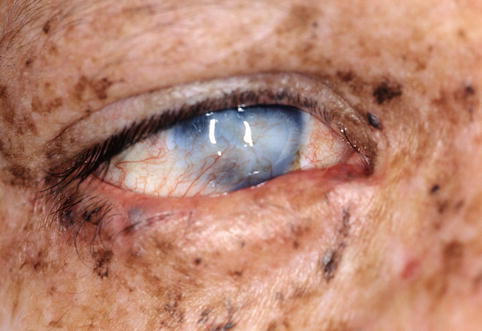
Fig. 11.3
XP – patient with xeroderma pigmentosum showing extensive cutaneous pigmentation and irregularities (Clinical picture courtesy of Dr George Bartley, Mayo Clinic)
11.4 Degeneration
11.4.1 Atrophy and Solar Elastosis
The aging skin (chronological) is characterized by atrophy of the epidermis with loss of elasticity [30]. The exposure to ultraviolet radiation (photodamage) in addition to the aging process, at least initially, results in a hypertrophic repair response with a thickened epidermis and increased melanogenesis. In the dermis, the actinic damage causes massive elastosis (deposition of abnormal elastic fibers), collagen degeneration, and twisted, dilated microvasculature. These latter changes are recognized as “solar elastosis” [31, 32].
Microscopically, the collagen in the dermis is altered with increased basophilia due to degeneration of both collagen and elastic fibers [33, 34] (Fig. 11.4). The overlying epidermis is usually atrophic with decreased number and size of rete pegs. Patches of increased melanocytes are seen in the basal layer of the epithelium [32, 35].
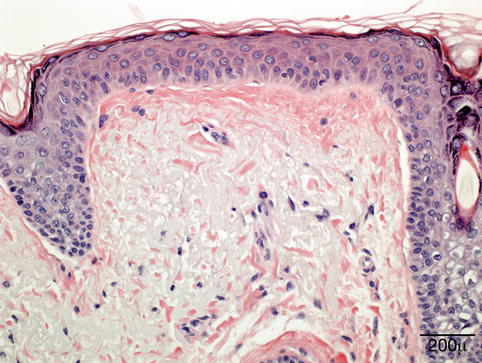
Fig. 11.4
Elastotic degeneration – eyelid biopsy shows in the superficial dermis fragmentation of the collagen and basophilic elastotic degeneration (hematoxylin and eosin stain; original magnification 400×)
In certain aging-associated conditions such as senile ectropion (turning out) and entropion (turning in), in addition to the changes described above, one might also find chronic inflammation and dermal scarring. In dermatochalasis (an aging change characterized by redundant upper eyelid skin, hanging in folds over the eye), histologically, the skin appears atrophic; and in the dermis, there is loss of collagen with basophilic degeneration, chronic inflammation, and increased numbers of capillary vessels [36].
11.5 Inflammation
11.5.1 Blepharitis
Definition
There is no stringent definition for blepharitis. In broader terms, blepharitis means inflammation of the eyelids as a whole. Marginal blepharitis refers to inflammation of the eyelid borders, although, in practice, “marginal” is usually omitted so that blepharitis is understood as inflammation of the eyelid border.
Epidemiology
Blepharitis is a common problem in ophthalmology practice, but the true incidence is unknown. It can affect any age group.
Etiology
Multiple pathomechanisms are involved in blepharitis [37]. Anterior blepharitis is often caused by overgrowth of coagulase-negative staphylococci in a patient with seborrheic dermatitis. An imbalance among various ocular bacterial species is also a possibility [38]. Posterior blepharitis is a consequence of meibomian gland dysfunction, conjunctival inflammation, or general conditions such as acne rosacea [39]. The role of Demodex mites (Fig. 11.5) in the etiology of blepharitis is controversial as they are present in healthy subjects as well as in patients with blepharitis, although, in the latter, their number is greatly increased [40].
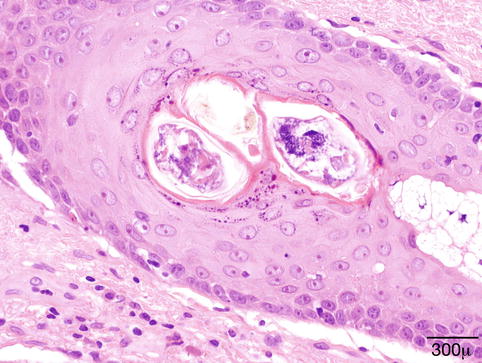
Fig. 11.5
Demodex folliculorum – Demodex mites in lash follicle. No evidence of inflammation (hematoxylin and eosin stain; original magnification 600×)
Clinical Features
Blepharitis is a chronic process. Anterior blepharitis affects the lid margin anterior to the gray line with squamous debris or collarettes around the base of lashes. Posterior blepharitis mostly manifests with problems related to meibomian gland dysfunction leading to tear film instability with irritation and discomfort. Telangiectasia of the eyelid border is a prominent feature of rosacea.
Macroscopy
The eyelids are red and swollen with flakes and collarettes around the base of the lashes (Fig. 11.6).
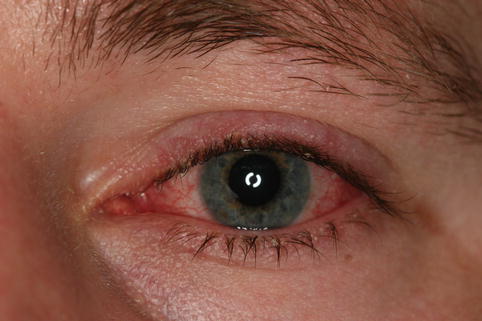
Fig. 11.6
Seborrheic blepharitis – hyperemic swollen eyelids, with telangiectasias and greasy scales on the lashes in young man
Histopathology/Immunoprofile
Biopsy is seldom performed for blepharitis unless the condition is unilateral and nonreacting to therapy, hence, suspicious for an underlying malignant condition diffusely infiltrating the lid.
Differential Diagnosis
Pagetoid infiltration of the eyelid epithelium by sebaceous gland carcinoma can mimic chronic blepharitis. The condition is unilateral.
Genetics
There is no known genetic predisposition to blepharitis.
Prognosis and Predictive Factors
Chronic blepharitis predisposes to the development of styes and chalazia and can lead to atrophy of the meibomian glands, loss of lashes, and trichiasis (misdirected growth of lashes). The latter is further complicated by ocular surface disease and corneal lesions.
11.5.2 Hordeolum (Stye)
Definition
An acute, clinically painful, purulent infection of the eyelid glands of Zeis (external hordeolum) or Meibom (internal hordeolum), mainly caused by staphylococcal bacteria
Epidemiology
The lesion is fairly common in both children and adults.
Clinical Features
Initially the eyelid is swollen, edematous, and erythematous; then a purulent papule becomes visible. Spontaneous or surgical drainage regularly solves the problem. Histopathological material is usually not submitted.
11.5.3 Chalazion
Definition
A granulomatous reaction to the sebaceous material of the meibomian glands (lipogranuloma)
Epidemiology
This is a very common lesion in young females (10–29 years) and older men (>60 years) [41]. Patients with posterior blepharitis or rosacea are prone to the development of chalazia.
Etiology
Stricture or obstruction of the duct of the meibomian gland leads to disruption of the gland, and lipid material/sebum is spilled into the surroundings and followed by a granulomatous reaction.
Clinical Features
Chalazion presents clinically as a bump in the tarsal plate protruding under the overlying, normal-looking skin. The conjunctival surface may harbor a pyogenic granuloma.
Macroscopy
The material from a curettaged chalazion is jellylike, yellowish gray, and friable. Lesions that extend anteriorly or posteriorly may clinically mimic a malignant tumor and may be excised.
Histopathology
Microscopy shows a classic granulomatous reaction composed of epithelioid macrophages, multinucleated giant cells, plasma cells, lymphocytes, and leukocytes, centered around empty spaces corresponding to dissolved lipid (Fig. 11.7).
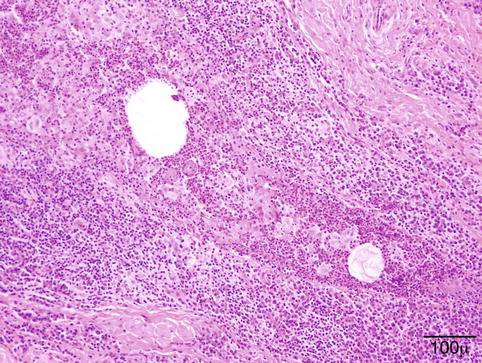
Fig. 11.7
Chalazion- granulomatous reaction with epithelioid macrophages and mixed inflammatory cells centered around dissolved lipid droplets (hematoxylin and eosin stain; original magnification 200×)
Differential Diagnosis
Other granulomatous reactions, tuberculous granuloma, sarcoidosis, cat scratch disease [42], insect bite
Prognosis
Very good, may resolve spontaneously, but has a tendency to recur in patients with blepharitis
11.5.4 Acne Rosacea
Definition
Rosacea (acne rosacea) is a common chronic inflammatory skin disease characterized by persistent erythema, telangiectasia, papules, and pustules. It affects mainly the midfacial zone and eyes.
Epidemiology
Rosacea is very common in fair-skinned people of northern ancestry; one-third have a family history of the disease. It mostly affects adults over 30 years of age [43] and shows a female predominance, although ocular rosacea affects both genders equally. It is probably one of the most underdiagnosed skin and ocular conditions.
Etiology
Rosacea reflects an abnormal sensitivity to the environment that is exacerbated by external triggers including UV light, heat, and a variety of microbes. There is an excessive activation of innate immune effector molecules, such as cathelicidin, an antimicrobial peptide, and kallikrein 5 (KLK5), the serine protease responsible for processing cathelicidin [44, 45]. Disturbed cathelicidin processing results in peptide fragments causing inflammation, erythema, and telangiectasia [46]. Infectious agents such as Demodex and Demodex-associated Bacillus oleronius [47–49] may act as triggers by stimulating overexpression of TLR2, which, in turn, enhances KLK5 production in keratinocytes [50].
Clinical Features
Ocular rosacea is characterized by blepharitis, telangiectasia of the eyelid border, recurrent chalazia, and conjunctival hyperemia.
Histopathology
Early lesions may show a mild perivascular and perifollicular lymphocytic infiltrate with telangiectasia (Fig. 11.8). In granulomatous rosacea, epithelioid histiocytes and giant cells are organized into tuberculoid granuloma [51].
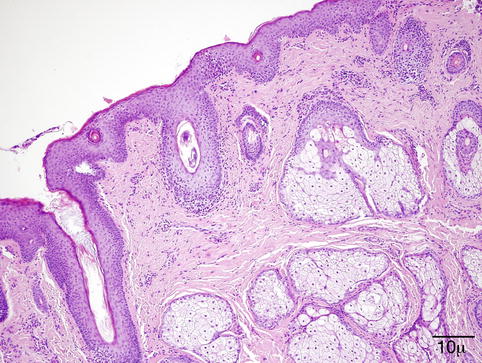
Fig. 11.8
Papular rosacea – perifolliculits with Demodex folliculorum and vascular dilation in early rosacea (hematoxylin and eosin stain; original magnification 100×)
Differential Diagnosis
Perioral dermatitis follows the use of topical steroids. Clinical correlation is needed to distinguish between the two entities. Granulomatous rosacea must be distinguished from sarcoid, granulomatous infection, and foreign body granuloma. Perifollicular granuloma speaks in favor of granulomatous rosacea [52].
Prognosis
Rosacea is a progressive condition; progression can be delayed by avoiding the triggers that provoke flare-ups (heat, UV radiation, alcohol).
11.5.5 Pyogenic Granuloma
Definition
Pyogenic granuloma (PG) is defined as an inflammatory and hyperplastic lesion by some authors [53] and a vascular tumor (lobular capillary hemangioma) by others; the terms are used interchangeably.
Epidemiology
PG is a very common lesion of the skin and mucous membranes and occurs most frequently in children and young adults.
Etiology
The etiology of PG is uncertain; it has been related to trauma, but a study of 178 cases disclosed a previous trauma in only 7 % [54]. Hormonal influence and/or drugs (retinoid, antiretroviral therapy, antineoplastic agents) have all been implicated in the pathogenesis.
Clinical Features
Clinically, the lesion presents as a rapidly growing, bright red polypoid papule or nodule that bleeds easily (Fig. 11.9).
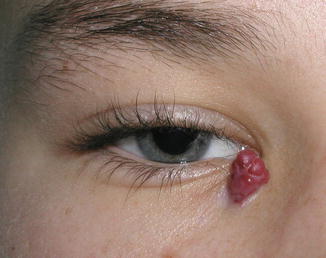
Fig. 11.9
Pyogenic granuloma – lobulated circumscribed purple tumor at the medial canthus in a young girl
Histopathology/Immunoprofile
Initially, PG is similar to granulation tissue, with capillaries and venules arranged in a radial pattern. The stroma is markedly edematous and infiltrated by a mixture of inflammatory cells (Fig. 11.10). Fully developed PG shows bland blood vessels proliferating in a lobular pattern, lined by plump endothelial cells. Inflammatory cells are few.
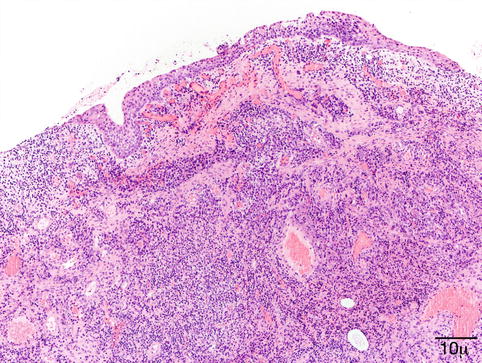
Fig. 11.10
Pyogenic granuloma – proliferating bland vessels, fibrosis, and intense inflammatory infiltrate are observed (hematoxylin and eosin stain; original magnification 100×)
Differential Diagnosis
Bacillary angiomatosis occurs mostly in HIV-infected patients and is usually multiple; the causative Bartonella species may be evidenced by Warthin–Starry modified silver stain or a monoclonal antibody [57].
Infantile hemangiomas express GLUT1 [58], which is negative in PG.
Prognosis
The prognosis is usually good. The best treatment option with the lowest recurrence rates is complete surgical excision. Laser therapy and intralesional corticosteroids also give good results.
11.5.6 Viral Diseases
11.5.6.1 Molluscum Contagiosum
Definition/Etiology
Molluscum contagiosum is a benign contagious lesion caused by a DNA poxvirus.
Epidemiology
Molluscum contagiosum is most common in children and young adults. It is spread by skin-to-skin contact or autoinoculation; sexual transmission occurs as well. Immunocompromised individuals are at increased risk.
Localization
This occurs mostly on the face and trunk. When located at or close to the eyelid border, follicular conjunctivitis is a complication.
Clinical Features
Molluscum contagiosum presents clinically as small, skin-colored, either solitary or multiple umbilicated papules (Fig. 11.11). The lesions may be more numerous, confluent, and larger in immunocompromised patients.
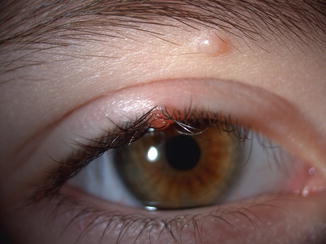
Fig. 11.11
Molluscum – a typical small umbilicated papule is present under the brow and another at the eyelid border in a young child
Histology
Molluscum contagiosum is composed of endophytic lobules of proliferating squamous epithelium, which expands into the dermis. The suprabasal epithelial cells contain large, faintly granular, eosinophilic cytoplasmic inclusions (Henderson–Patterson or molluscum bodies) that displace the nucleus. The molluscum bodies increase in size, become more basophilic, and are finally extruded through a central ostium onto the surface of the skin (Fig. 11.12).
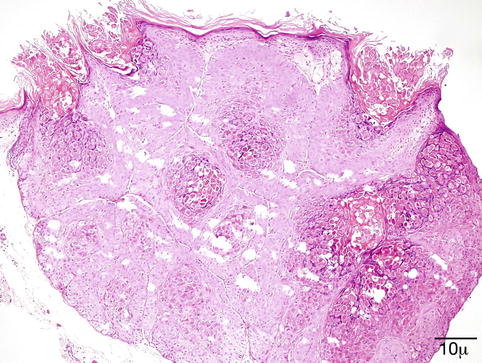
Fig. 11.12
Molluscum – endophytic epithelial lobules and characteristic intracytoplasmic eosinophilic inclusion bodies are conspicuous. Molluscum bodies are also extruded onto the surface (hematoxylin and eosin stain; original magnification 100×)
Differential Diagnosis
The intracytoplasmic eosinophilic inclusions are pathognomonic.
Prognosis
In immunocompetent patients, the lesion may spontaneously involute within 6–12 months. Recurrence is rare whether treated or not.
11.5.6.2 Verruca Vulgaris
Definition
Verruca vulgaris is a benign, rough-surfaced growth caused by certain human papillomavirus strains (HPVs 1, 2, 4).
Epidemiology
Verruca vulgaris is very common; it affects mostly children and adolescents. An increased frequency is noted in immunocompromised patients. Warts are relatively rare on the eyelids.
Clinical Features
The lesion presents as a papillomatous, exophytic papule with hyperkeratosis (Fig. 11.13).
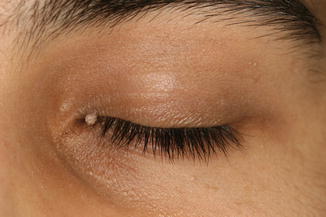
Fig. 11.13
Verruca vulgaris – small skin-colored wart on the upper eyelid of a teenager
Histopathology
The surface is hyperkeratotic. The epidermis is papillomatous and acanthotic. The granular cell layer is prominent with coarse keratohyalin granules. Koilocytes are present in the upper layers of the epidermis. The rete ridges are elongated and tend to bend inward (Fig. 11.14).
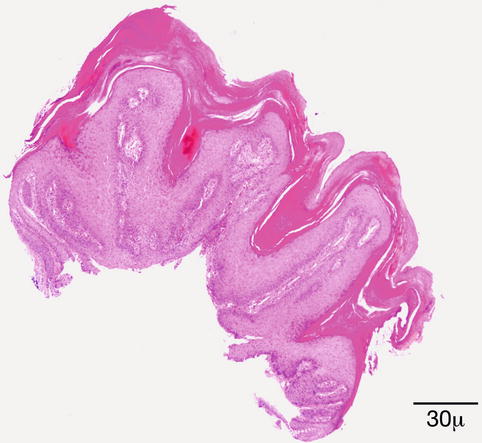
Fig. 11.14
Verruca vulgaris – the lesion is papillomatous; epidermal hyperplasia and hyperkeratosis are present. The rete ridges are elongated and point towards the center (hematoxylin and eosin stain; original magnification 40×)
Differential Diagnosis
The most important differential is from squamous cell carcinoma. Full-thickness atypia and loss of cellular polarity are not seen in verrucae. Actinic keratosis and seborrheic keratosis may have a verruca-like appearance.
Prognosis
Warts may regress spontaneously; however, despite the great variety of treatment options, recurrences are frequent. In immunocompromised patients, the lesions may be widespread and even more resistant to treatment.
11.5.6.3 Herpesvirus Infection
Definition
Infection, primary or recurrent, caused by the herpes simplex DNA virus (HSV).
Synonyms
Cold sores
Epidemiology
The distribution of HSV infections is worldwide; 65–90 % of the population shows seropositivity for HSV1 or HSV2. The infection is generally acquired during childhood from an infected person who sheds the virus. The virus enters through the skin or mucosa.
Localization
The primary infection mainly affects the oral mucosa, rarely the conjunctiva; however, recurrences are more common in the eye. The actively replicating virus follows the course of the sensory nerves and reaches a sensory ganglion (trigeminal, superior cervical, or ciliary), where it establishes latency.
Clinical Features
Primary infection of the eye may appear as a blepharitis with vesicles/blisters on the eyelid margin, accompanied by conjunctivitis. In about two-thirds of cases, corneal lesions also develop [59]. Recurrent lesions may also take the form of blepharitis or blepharoconjunctivitis, followed several days later by corneal manifestations. In immunosuppressed or HIV-positive patients, the eyelid lesion may become ulcerated [60] or may even mimic a tumor [61, 62].
Histopathology
Early changes are noticed in the epidermal cell nuclei. The nucleus swells and takes on a ground-glass appearance, and the chromatin forms peripheral clumps. There is vacuolization of the cytoplasm first in the basal cell layer; but soon, the entire epidermis is involved, followed by acantholysis and intraepidermal vesicle formation. The cytoplasm of these cells becomes eosinophilic and homogeneous. Some of the cells are multinucleated (Tzanck cells). Eosinophilic intranuclear inclusions may be seen in the multinucleated cells. In the dermis, there is a perivascular lymphohistiocytic inflammation.
Differential Diagnosis
Typical blistering lesions caused by HSV1 or HSV2 are diagnosed clinically. However, lesions mimicking tumors in HIV-positive patients may be biopsied to reach a diagnosis. The inflammatory reaction is granulomatous; nuclear inclusions consistent with HSV may be seen. Culture or PCR may eventually confirm the diagnosis.
Prognosis and Predictive Factors
Although the clinical manifestation is recurrent in a proportion of cases, with time the interval between attacks becomes progressively longer. Corneal complications may lead to severe visual impairment.
11.5.7 Bacterial Infections
Infections with Streptococcus pyogenes (group A Streptococcus) range from superficial impetigo to deeply invasive disease such as necrotizing fasciitis. Staphylococcus aureus may participate in a combined infection [63], especially in cases of necrotizing fasciitis [64]. It seems that the predominant infectious agent varies with geographic regions and socioeconomic level.
Synonyms
Pyoderma, school sores
Epidemiology
Impetigo occurs most commonly in economically disadvantaged, preschool-aged children.
Localization
Mostly seen in the perioral area and on the nose, sometimes affects the eyelids as well
Clinical Features
Minor trauma, bites, or burns of systemic disease may be elicited from the clinical history. Impetigo starts as a papule, quickly evolving into a vesicle with perivesicular erythema. The vesicle turns into pustules that gradually enlarge and break down, forming thick scabs [65]. Bullous impetigo occurs in nontraumatized skin and is characterized by blisters or moist erosions. Necrotizing fasciitis affects the skin and deep soft tissues.
Histopathology
Impetigo is characterized by the development of a subcorneal pustule beneath the granular layer. Gram-positive cocci may be found within the pustule or in neutrophil granulocytes. The dermis shows perivascular infiltration by neutrophil granulocytes and lymphocytes. Thrombosis of the microcirculatory vessels in necrotizing fasciitis is the pathogenic background for gangrene.
Differential Diagnosis
The diagnosis is usually clinical. Secondarily infected pemphigus and pustular psoriasis seldom affect the eyelids.
Prognosis and Predictive Factors
Impetigo can resolve spontaneously or with local/oral antibiotic treatment. Post-streptococcal glomerulonephritis is a rare complication. Necrotizing fasciitis may be fatal in 10 % of cases; intensive treatment with surgical debridement of affected tissues is mandatory.
11.5.8 Fungal and Parasitic Infections
Definition
Fungal infection of the eyelid can be primary, the consequence of overgrowth of the commensal flora of the eyelid skin, in case of local or systemic immune depression, or direct inoculation into the skin. Another possibility is that the eyelid infection is the manifestation of hematogenous spread of a fungal infection. Parasitic infections are mostly the result of transmission by a vector, e.g., a fly, mosquito, or tick.
Epidemiology
Some fungi and parasites have a worldwide distribution; others are endemic to certain areas. Hence, the geographic location is a significant determining factor in the development of fungal or parasitic infections [66].
11.5.9 Fungal Infections
11.5.9.1 Histoplasmosis
Histoplasma capsulatum is endemic in North America. Cutaneous manifestation is usually only seen in immunocompromised individuals. The histopathological features vary. There is a dermal infiltrate with foamy histiocytes, granulomatous infiltration, and a variable amount of inflammatory cells. The organism appears intra- or extracellularly such as small yeastlike structures within a clear halo.
11.5.9.2 Cryptococcosis
Cryptococcus neoformans has a worldwide distribution. The eyelid cutaneous manifestations are usually part of a systemic disease in immunocompromised patients [67] but can also occur as a primary skin disease. There is a dense dermal infiltrate of foamy histiocytes with or without a granulomatous inflammation. Mucin stains, PAS, and Grocott highlight the thick capsule of the pathogen.
11.5.9.3 Sporotrichosis
Sporotrichosis is caused by the dimorphic fungus Sporothrix schenckii. It can present with a disseminated, lymphocutaneous, or cutaneous form. Transmission usually occurs via direct implantation in the skin. The solitary cutaneous form presents as a verrucous or ulcerated nodule. Histology shows a suppurative granuloma; PAS or Grocott stains of multiple serial sections are usually needed to demonstrate the 4–6 μm spores.
11.5.9.4 Blastomycosis
Blastomycosis is endemic in North America. Blastomyces dermatitidis is a dimorphic fungus. The inhaled spores transform into yeast and spread hematogenously. The most common extrapulmonary involvement is the skin [68]; eyelid involvement is uncommon [69]. Rarely direct inoculation is followed by a primary cutaneous lesion, which is verrucous or ulcerated. Histologically, the picture closely resembles sporotrichosis. The skin surface shows pseudoepitheliomatous hyperplasia. Fungal spores are thick walled and exhibit a broad-based budding, which helps in its identification.
11.5.10 Parasitic Infections
Leishmaniasis (oriental sore) is a parasitic infection caused by protozoa of the genus Leishmania, transmitted by sand fly. Leishmaniasis is endemic in tropical and subtropical areas and the Mediterranean countries. Recently a significant number of leishmaniasis cases have been reported in travelers [70] to endemic countries; thereupon, this led to an increased awareness of the condition. Leishmaniasis in travelers is characteristically the cutan type [71]. The mucocutaneous type is generally associated with infection acquired in Latin America [72].
In immunocompetent patients, the infection is usually self-limited. It is rare in the eyelid; the clinical presentation can simulate a cutaneous appendage tumor [73], a basal cell carcinoma (BCC) [74], or, more commonly, a chalazion [75]. The typical periocular location is the lateral canthus [76]. The histopathological picture depends on the time of biopsy and on the host response. In the early stages, the presentation mimics a xanthogranulomatous inflammation; macrophages aggregate in the dermis. Later, epidermal hyperplasia develops with ulceration. A dense dermal infiltrate of histiocytes, plasma cells, and lymphocytes is seen. The inflammatory infiltrate later becomes granulomatous. The Leishmania amastigotes are recognized in the cytoplasm of histiocytes (Leishman–Donovan bodies) as round organisms seen with hematoxylin and eosin (H&E) or Giemsa stain, as a rule, in the non-granulomatous areas. Recently, polymerase chain reaction (PCR) is also available for diagnostic purposes.
11.5.11 Filarial Infections
Dirofilaria repens is endemic in the Mediterranean area; the reservoirs are dogs or cats, and the larvae are accidentally transmitted to humans by bite of mosquito vectors. It has been reported with increasing frequency from areas outside the endemic territories [77]. The larva cannot survive in humans but is able to wander under the conjunctiva to the orbit, through the bridge of the nose from one eye to the other (personal observations), or to the lacrimal gland [78]. In the eyelid, the infection presents as an anterior cellulitis not responding to conventional therapy and is generally biopsied (Fig. 11.15). The filaria provokes a granulomatous inflammatory reaction with foreign body giant cells and a markedly eosinophilic infiltrate (Fig. 11.16). The worm is recognized by its thick ridged cuticle and well-developed musculature (Fig. 11.17). As there is no blood filariasis, no systemic therapy is necessary.
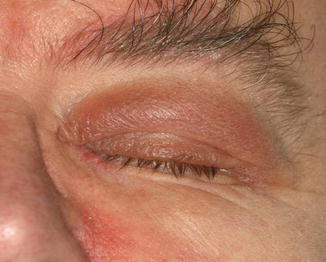
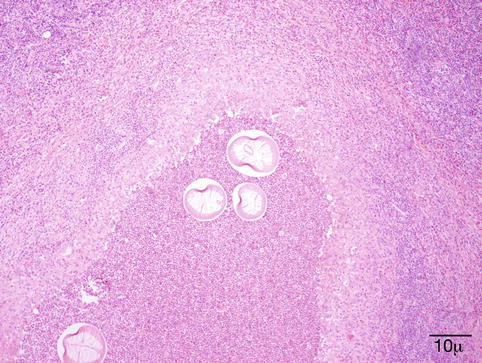
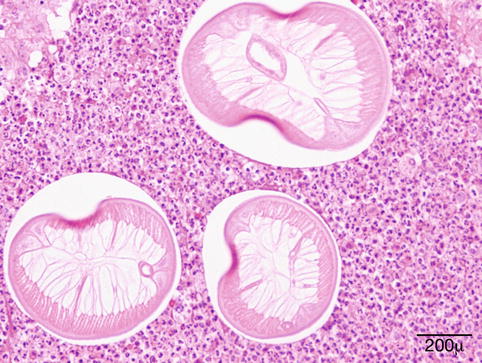
Fig. 11.15
Dirofilaria repens – the elderly male patient had an anterior cellulitis that did not react to conventional therapy. Biopsy disclosed a granulomatous inflammation caused by D. repens (Clinical picture courtesy of Dr. Olga Lukáts, Semmelweis University, Dept. of Ophthalmology)
Fig. 11.16
Dirofilaria repens – an intense granulomatous reaction with foreign body giant cells and numerous eosinophils is present around cross sections of a worm (hematoxylin and eosin stain; original magnification 100×)
Fig. 11.17
Dirofilaria repens – the multilayered ridged cuticle, thick musculature, and the presence of uterus help recognition of a female worm (hematoxylin and eosin stain; original magnification 400×)
Loa loa is a nematode endemic in the rain forest region of West and Central Africa. The nematodes are transmitted by Chrysops flies. Many patients remain asymptomatic for years after the infection. Loa loa has a predilection for the conjunctiva and periocular tissues [79, 80]. In an extensive review of the literature on loaiasis in Europe and the United States from 101 cases reported, 48 experienced eye worm migration [81]. The clinical history provides important clues to the diagnosis (travel to or immigration from endemic countries). The filaria can be demonstrated in blood smears or by PCR.
11.6 Eyelid Lesions with Association to Systemic Diseases
A few lesions occurring in the eyelid region might be associated with systemic disorders. In fact, on occasion, the eyelid lesion might be the first manifestation of disease, hence the importance for both ophthalmologists and ophthalmic pathologists to be aware of these.
11.6.1 Xanthelasma
Definition
It is a form of cutaneous xanthoma, a group of lesions characterized by the accumulation of lipid-rich macrophages also known as foamy cells [82]. These lesions are usually associated with disorders of lipoprotein metabolism, although only a minority of individuals with such disorders develop xanthomas [82, 83]. Therefore, an increased vascular permeability has been implicated in the pathogenesis of these lesions [84].
Clinical Features
The eyelid and periocular skin are the most affected sites. Clinically, xanthelasma presents as yellowish, noninflamed flat or slightly elevated plaques originating in the medial eyelid and spreading laterally in a symmetric fashion (Fig. 11.18). These are not painful and do not ulcerate the skin. Xanthelasma is more commonly seen in the Asian and Mediterranean populations.
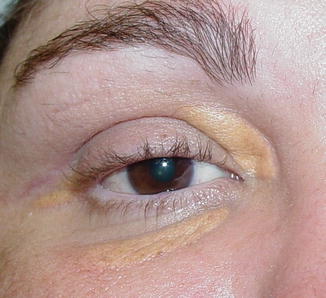
Fig. 11.18
Xanthelasma – It presents clinically as yellowish, non-inflamed flat plaques originating in the medial eyelid and spreading laterally (Clinical picture courtesy of Dr James Garrity, Mayo Clinic)
In approximately 50 % of the patients, these lesions are associated with elevated plasma lipid levels. These might be due to a hereditary genetic abnormality such as dyslipoproteinemia, familial hypertriglyceridemia, familial lipoprotein lipase deficiency, or hyperlipidemias, secondary to diabetes mellitus type II [84–87].
Histological Features
Microscopically, the dermis is infiltrated diffusely by numerous foamy histiocytes, forming perivascular aggregates (Fig. 11.19). The overlying epidermis is not affected. Areas of necrobiosis, vasculitis, or a prominent associated lymphoid infiltrate should not be seen, and the presence of these findings should evoke other diagnoses such as necrobiotic xanthogranuloma.
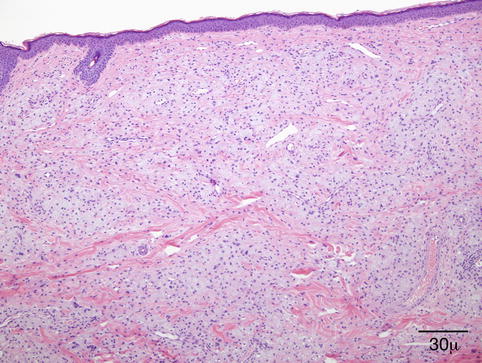
Fig. 11.19
Xanthelasma – Microscopically, it is characterized by a diffuse dermal infiltration of foamy histiocytes (hematoxylin and eosin stain; original magnification 40×)
On immunohistochemistry, the foamy cells are positive for macrophage markers such as CD68 and CD163 and negative for S-100 and CD1a. Because the differential diagnosis also includes lesions with clear cell morphology such as balloon cell melanoma and balloon cell nevus, melanocytic markers might be helpful.
Treatment
Xanthelasma in most patients requires no treatment. Lesions might be treated for cosmetic reasons. Various treatments have been studied, including surgical excision, treatment with chemicals, and ablative laser therapy; but these methods have disadvantages. Recently, nonablative laser therapy has been proposed as a treatment for xanthelasma palpebrarum [88].
11.6.2 Calcinosis Cutis
Definition
The term calcinosis cutis is used to describe a group of diseases characterized by calcium deposits in the skin. Historically, these have been divided into dystrophic type when calcium deposits occur in previously damaged tissue and the less common metastatic form, which is associated with elevated serum calcium and/or phosphate levels [89]. In many cases, the pathogenic mechanism is not identified, and these cases have been assigned as idiopathic. Herein, we will discuss the most common form occurring in the eyelid region, the subepidermal calcified nodule.
Pathogenesis
The pathogenesis is not completely understood. Insoluble compounds of calcium are deposited within the skin, primarily composed of hydroxyapatite crystals or amorphous calcium phosphate, with no known predisposing factors. Although systemic associations, such as hyperparathyroidism, renal insufficiency, and other disorders of calcium/phosphate, have been described in metastatic calcinosis and an underlying tissue abnormality described with dystrophic calcification [90], an underlying systemic or local cause has not been confirmed in association with subepidermal calcified nodules [91].
Clinical Presentation
Subepidermal calcified nodule usually occurs as a small, <5 mm, solitary, white-yellowish nodule on the eyelid of young children in their first or early second decade of life [91, 92] (Fig. 11.20). Multiple lesions can be seen but are less frequent [93].
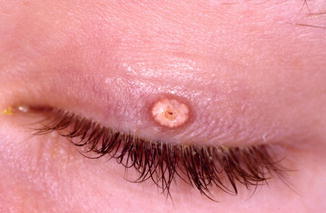
Fig. 11.20
Calcinosis cutis – solitary white-yellowish nodule in the upper eyelid of a young child (Clinical picture courtesy of Dr. George Bartley, Mayo Clinic)
Clinically, these might be mistaken for eyelid cyst, molluscum contagiosum, cutaneous horn, or a neoplasm in an older patient [91].
Histopathology
The characteristic finding is the presence of subepithelial, sharply demarcated basophilic amorphous deposits surrounded by variable amounts of histiocytic foreign body giant cell reaction. The overlying epidermis is usually acanthotic and papillomatous with occasional hyperkeratosis (Fig. 11.21). The deposits stain positive with von Kossa stain for calcium, and the histiocytes are positive for CD68 immunostain [91, 92, 94].
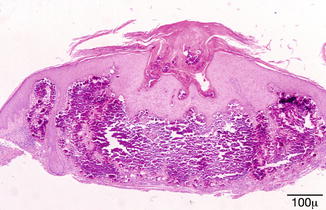
Fig. 11.21
Calcinosis cutis – dermal deposit of amorphous basophilic granular material surrounded by histiocytic foreign body reaction. The overlying epidermis shows acanthosis and hyperkeratosis (hematoxylin and eosin stain; original magnification 200×)
11.6.3 Sarcoidosis
Definition
Sarcoidosis is a multisystem disease of unknown etiology characterized by noncaseating granulomas in various organs. Granulomas most often appear in the lungs (85 %) and lymph nodes (34 %), but virtually any organ may be involved including the eye (27 %) and, less commonly, the central nervous system (1 %).
Epidemiology
It affects most commonly young adults of both sexes. Incidence is highest for individuals younger than 40 years and peaks in the age group from 20 to 29 years; a second peak is observed for women over age 50 years. The disease is most prevalent in Northern European countries. In the United States, sarcoidosis is more common in African descendant than Caucasians, with an annual incidence reported of 35.5 and 10.9/100,000, respectively [95–97].
Pathogenesis
The pathogenesis of this disease is not yet known. The most accepted hypothesis is that in genetically susceptible individuals, sarcoidosis is caused through alteration in immune response after exposure to an environmental, occupational, or infectious agent. Studies have shown an increase in B-cell activity with hypergammaglobulinemia in about 50 % of the patients. Reduced delayed hypersensitivity is also found in many patients. Immune dysregulation due to persistent low virulence antigen that is poorly cleared by the immune system, leading to a chronic T cell of the Th1 subtype response with granulomatous response, has been hypothesized. Medications that increase Th1 response, such as interferon, have been reported to trigger or exacerbate sarcoidosis. GLI1 oncogene has been found to be highly expressed in patients with sarcoidosis [98].
Clinical Features
The eye may be affected by sarcoidosis in a variety of ways, with the most frequent site being the uveal tract [99–101]. Optic nerve and optic nerve head involvement has been reported in about 5 % of patients with sarcoidosis. Sarcoid may involve the lacrimal gland and rarely extends into the adjacent orbital soft tissue, which was observed in only 2 of 202 cases (1 %) studied by Obenauf et al. [101].
Eyelid involvement is extremely rare, with only a few case reports in the literature [102–105]. Clinically, the lesions appear as slightly elevated, grayish-white, mildly umbilicated papules and nodules, some of which might become confluent (Fig. 11.22). Eyelid sarcoidosis may be an isolated manifestation of sarcoidosis or associated with systemic more often than ocular disease [102, 104, 105].
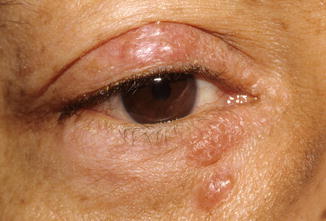
Fig. 11.22
Sarcoidosis – multiple eyelid elevated confluent nodules, grayish white with mild umbilication in areas (Clinical picture courtesy of Dr George Bartley, Mayo Clinic)
The diagnosis of sarcoidosis is often a matter of exclusion of other diseases including neoplasm. It is made on the basis of signs and symptoms, patient’s age, race, and laboratory findings. A definitive diagnosis requires a biopsy to be performed.
Histopathology
The typical lesion in the biopsy is characterized by circumscribed epithelioid granulomas with little or no necrosis and few Langhans giant cells (Fig. 11.23). Giant cells may contain asteroid or Schaumann bodies. A sparse lymphocytic infiltrate may be seen in the periphery of these granulomas, which is the reason these are referred to as “naked granulomas.”
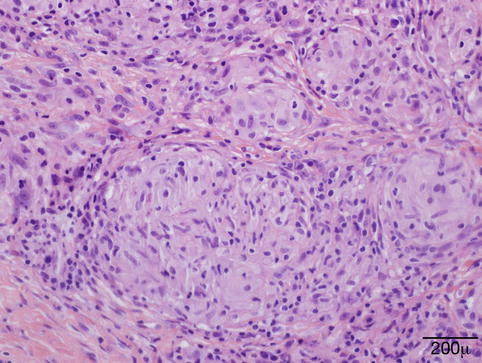
Fig. 11.23
Sarcoidosis – the typical sarcoid lesion is characterized by circumscribed epithelioid granulomas with little or no necrosis and few Langhans giant cells (hematoxylin and eosin stain; original magnification 400×)
Differential Diagnosis
This includes mainly granulomatous infectious processes such as tuberculosis and fungal infection. Therefore, special stains and tissue cultures for microorganisms must be obtained on a routine basis.
Prognosis
The course of the disease is variable, ranging from self-limited acute disease to a chronic debilitating disease that could result in death. Spontaneous remissions occur in two-thirds of the patients, but 10–30 % of patients have a more chronic or progressive course [106]. The mortality rate is 1–6 %. Death can be due to pulmonary fibrosis leading to respiratory failure, myocardial involvement leading to cardiac arrhythmias and cardiac failure, central nervous system (CNS) involvement, renal insufficiency, and liver disease [107].
11.6.4 Amyloidosis
Definition
Amyloidosis refers to a group of diseases characterized by the extracellular deposition of amorphous, eosinophilic hyaline material, constituted by insoluble fibrous protein aggregates of autologous origin in the connective tissue and perivascular spaces of various organs [108]. Clinically, amyloidosis is divided in five groups: primary systemic, primary localized, secondary to a chronic illness, secondary to multiple myeloma, and familial type. The involvement of the eyelids occurs in the first two forms.
Epidemiology
Primary systemic amyloidosis occurs in approximately 8 per million people per year, with approximately 3,000 new patients annually in the United States [109]. Median age at diagnosis is 64 years, but patients can present at any age. The male-to-female ratio is nearly 2:1. The prevalence of amyloidosis seems to be higher in the black and lower in the Asian populations, and it mirrors the prevalence of multiple myeloma and monoclonal gammopathies in the same populations. The incidence of secondary amyloidosis in the United States has been falling over the decades and accounts for only 3 % of systemic amyloidosis.
Etiology
The etiology of amyloidosis depends on its form. The causes for primary amyloidosis are still not well known, but it seems related to the presence of immunoglobulin free light chain circulating in the blood [110, 111]. The pathogenesis of amyloidosis associated to multiple myeloma and monoclonal gammopathies is better understood and has been the subject of extensive research [112–114].
Clinical Features
The skin of the eyelids is involved in either primary systemic or localized amyloidosis. Typically, the lesions are symmetrical, bilateral, single, or multiple, confluent yellowish or waxy papules [115]. Purpura is a common manifestation caused by vascular wall fragility, the result of amyloid deposits within the vessel walls (Fig. 11.24). Hemorrhagic papules that affect the eyelids have been considered diagnostic of systemic amyloidosis [116, 117]. Some patients might present with nonspecific chronic eyelid swelling, which might cause delay in the correct diagnosis [118, 119].
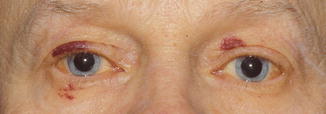
Fig. 11.24
Amyloid – some patients present clinically with hemorrhagic papules. These lesions have been considered highly indicative of systemic amyloidosis (Clinical picture courtesy of Dr. James Garrity, Mayo Clinic)
Histopathology
Papular lesions are the result of amyloid deposits in the superficial dermis. Clefting is often seen around large deposits. The amyloid also accumulates around and within vascular walls in the dermis and subcutaneous tissue (Fig. 11.25). This finding is observed in hemorrhagic lesions. Amyloid deposits may also be seen around cutaneous adnexa. No or scanty inflammation is observed in association with the amyloid deposits.
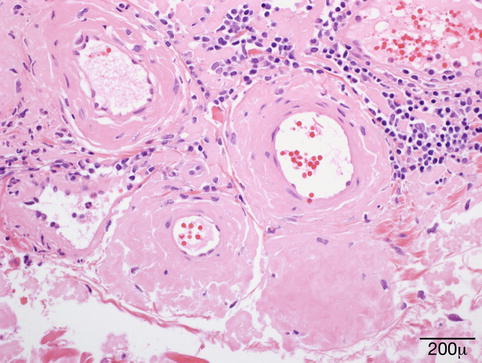
Fig. 11.25
Amyloid – eyelid biopsy shows dermal, predominantly perivascular deposits of amorphous eosinophilic hyaline material (hematoxylin and eosin stain; original magnification 400×)
Amyloid stains pink with hematoxylin and eosin and metachromatically with crystal violet and methyl violet [120]. With Congo red stain, amyloid is orange in color and displays an apple-green birefringence under polarized light (dichroism) (Fig. 11.26). Amyloid has a bright yellow-green color with Thioflavin T stain.

Fig. 11.26
Amyloid – orange in color with Congo red stain, under polarized light, however, amyloid shows an apple-green birefringence, also known as dichroism (hematoxylin and eosin stain; original magnification 200×)
A number of immunostains were developed to identify the protein components of amyloid and had been used to further classify amyloidosis. Currently, this has been largely replaced by subtyping, using tandem mass spectrometry analysis on formalin-fixed, paraffin-embedded specimens, due to the superior sensitivity and specificity of this latter technique [121].
Prognosis
The prognosis of amyloidosis involving the eyelids depends on the associated underlying disease. If the lesion is localized, surgical resection is the proposed treatment [122].
11.6.5 Juvenile Xanthogranuloma
Definition
It is a form of non-Langerhans cell histiocytosis, classified as World Health Organization (WHO) type II among a group of diseases known as histiocytosis. It is a benign, self-limited condition, rarely associated with systemic manifestations and typically occurring in the early childhood.
Epidemiology
Juvenile xanthogranuloma (JXG) is the most common of the non-Langerhans cell histiocytosis [123], although it is relatively rare. It had a reported incidence of 0.52 % among all pediatric lesions of the Kiel Pediatric Tumor Registry (KPTR) between 1965 and 2001 [124]. JXG may be present at birth; however, most lesions are diagnosed in the first year of life [124, 125]. There is a slight male predominance [124].
Etiology
The cause of JXG is unknown. JXG is a type of non-Langerhans cell histiocytosis, which derives from CD34+ stem cells that differentiate into CD14+ dermal/interstitial dendrocytes, considered to be the precursor cells for JXG [123].
Clinical Presentation
Clinically, it presents as single or multiple lesions, firm and slightly raised papulonodules measuring a few millimeters (Fig. 11.27). These may vary in color from reddish or yellowish to brown. The lesions can occur in any location and are most common on the head/neck and upper trunk. The lesions are usually solitary, but multiple lesions and extracutaneous systemic involvement may occur, such as the liver, lung, spleen, lymph nodes, bones, and gastrointestinal tract [125, 126].
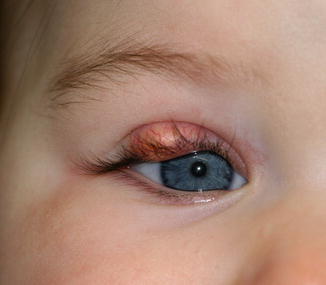
Fig. 11.27
JXG – small yellowish nodule in the eyelid of a young child. The overlying skin is smooth. No signs of inflammation (Clinical picture courtesy of Dr. Elizabeth Bradley, Mayo Clinic)
JXG may involve the skin of the eyelid and less commonly intraocular tissues, such as the iris, ciliary body, cornea, and episclera. Juvenile xanthogranuloma is known to cause spontaneous hyphema if the lesion involves the iris. Other ocular clinical presentations may include uveitis, heterochromia iridis, or an iris mass.
Histopathology
Microscopically, JXG is characterized by a dense dermal and subcutaneous tissue histiocytic infiltrate predominantly containing variable number of Touton giant cells (Fig. 11.28). The latter are multinucleated giant cells with a ring or wreath of nuclei surrounded by foamy cytoplasm and considered pathognomonic of JXG. Spindle cells can also be seen in the background. Other inflammatory cells such as lymphocytes, eosinophils, neutrophils, and mast cells are rarely present. The infiltrate surrounds the preexisting cutaneous adnexa, and the overlying epidermis is usually normal or thinned. Mature lesions tend to be more lipidized, and regressing lesions may show a prominent fibrous background [127].
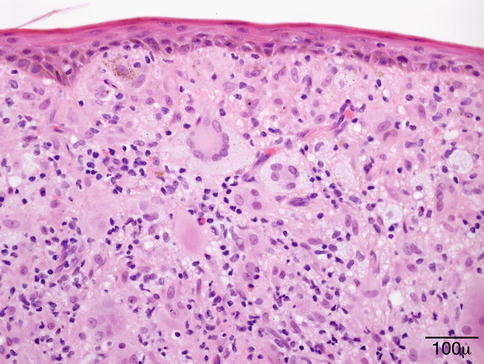
Fig. 11.28
JXG – dense dermal histiocytic infiltrate containing numerous Touton-type giant cells. These are characterized by a ring of nuclei surrounded by foamy histiocytes. A few eosinophils are seen intermixed (hematoxylin and eosin stain; original magnification 200×)
Immunostaining is important in establishing the diagnosis. JXG stains positive for factor XIIIa, CD68, CD163, CD14, and fascin. S-100 and CD1a, specific for Langerhans cells, are usually negative [124, 125].
The differential diagnosis includes Langerhans cell histiocytosis, which is positive for CD1a and S-100 immunostains, xanthoma, and less commonly dermatofibroma. These latter two lesions are discussed below.
Treatment and Prognosis
Cutaneous JXG usually regresses spontaneously and requires no treatment. Ocular JXG should be managed by an ophthalmologist, and the required treatment will depend on type of ocular involvement and complications.
Symptomatic systemic lesions, often due to a mass effect, may require treatment if they do not regress spontaneously [126]. Treatment may involve excision, radiation, or chemotherapy [128].
In general, the prognosis is excellent and disease-related deaths are uncommon but can typically occur in younger children with central nervous system or massive hepatic involvement [129].
11.6.6 Necrobiotic Xanthogranuloma
Definition
Necrobiotic xanthogranuloma (NXG), a distinctive dermal and subcutaneous xanthogranulomatous reaction, is a rare form of paraneoplastic syndrome often associated with monoclonal paraproteinemia [130] or other hematologic diseases. It is potentially a disfiguring disease that can involve the face, trunk, or extremities with a particular predilection for the periorbital region.
Epidemiology
The prevalence of disease is slightly higher in women than men, and onset is usually in the sixth decade of life (mean age 59.76 years).
Pathogenesis
The pathogenesis of NXG remains unknown. Because of the strong association with paraproteinemia, the causative role of this abnormal immunoglobulin has been speculated. It was thought that perhaps the increased serum immunoglobulins would combine with lipids and be deposited in the skin, leading to a foreign body giant cell reaction and the characteristic histopathological features of necrobiotic xanthogranuloma [131]. Others have thought that the activation of monocytes in vivo may contribute to the intracellular accumulation of lipoprotein-derived lipids, leading to non-inherited xanthomatosis [132, 133].
Clinical Features
Clinically, NXG is characterized by large, often yellow, indurated plaques sometimes associated with telangiectasia (Fig. 11.29). The lesions often become inflamed, leading to superficial ulceration. The most common locations are the periorbital region and thorax. Extensive lesions involving the eyelids may extend into the orbital soft tissues.
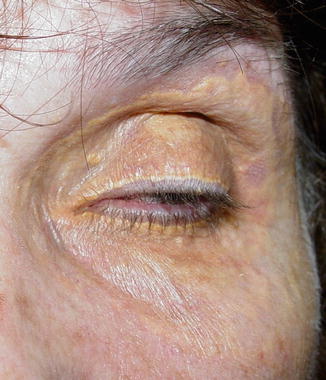
Fig. 11.29
NXG – clinical picture shows yellow, indurated plaques in both eyelids (Clinical picture courtesy of Dr. James Garrity, Mayo Clinic)
The clinical differential diagnosis includes mainly xanthelasma and xanthoma, lesions that are not usually indurated. The association of NXG with monoclonal gammopathy is very characteristic. Although IgG monoclonal gammopathy represents the most common association [134–137], a rare form of IgA gammopathy has also been reported [138], as well as the association with other hematologic disorders such as multiple myeloma, chronic lymphocytic‑leukemia, lymphoma, and other less common diseases such as cryoglobulinemia.
Histopathology
The histopathology is characterized by infiltrates containing mostly macrophages, foamy cells associated with extensive necrobiosis (degeneration of collagen) surrounded by palisading granulomatous reaction (Fig. 11.30). Numerous giant cells, Touton and foreign-body types, are usually seen. Cholesterol clefts, which have been reported previously, do not appear to be as common as initially thought [134]. Other findings include lymphoid follicles and a mixture of other inflammatory cells.
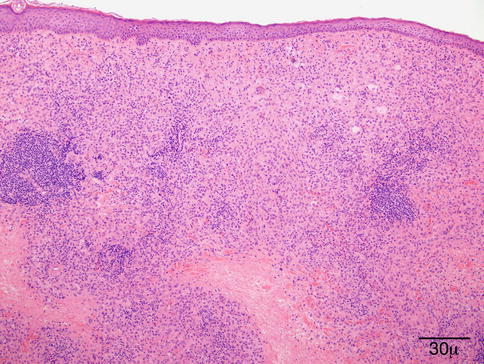
Fig. 11.30
NXG – eyelid biopsy shows dense dermal infiltrate containing mostly macrophages, Touton-type giant cells, and extensive areas of necrobiosis, surrounded by histiocytic granulomatous reaction (hematoxylin and eosin stain; original magnification 40×)
The differential diagnosis includes other necrobiotic disorders, especially necrobiosis lipoidica, subcutaneous granuloma annulare, and rheumatoid nodule. In NXG, the necrobiosis is more extensive than in other diseases and often occurs in broad bands. More important, the clinical presentation differs in each of these lesions. Necrobiosis lipoidica tends to affect mainly pretibial areas in diabetic patients. Granuloma annulare is characterized by papules often in young adults, and rheumatoid nodules are seen in patients with clinically apparent rheumatoid arthritis. Other lesions considered in the differential because of the presence of foamy histiocytes and Touton-type giant cells are xanthogranuloma and Erdheim–Chester disease. These entities do not show the necrobiosis that characterizes NXG.
Prognosis
Necrobiotic xanthogranuloma can cause impairing sequela. The cutaneous lesions tend to progress, and the course is unpredictable with periods of stabilization and reduction and more aggressive periods with extensive ulceration [135, 139]. The treatment is difficult and should include management of the underlying systemic disease and control of the skin lesions. Surgical removal of the lesions is generally not recommended because of the risk of recurrence, which was around 42 % in a large series [135]. However, surgery might be necessary to correct scarring and eyelid malfunction. Systemic prednisone and steroid injections have been proposed as alternative forms of treatment as well as low-dose radiation therapy and alkylating regimens.
11.6.7 Granulomatosis with Polyangiitis (Wegener’s Granulomatosis)
Definition
Wegener’s granulomatosis (WG), recently proposed to be renamed granulomatosis with polyangiitis (GPA) [140], is an uncommon disease defined by vasculitis involving small vessels, extensive “geographic” necrosis, and granulomatous inflammation classically involving the kidneys (glomerulonephritis) and the upper and lower respiratory tract (sinuses, nose, trachea, and lungs). The spectrum and severity of disease is variable, ranging from an indolent course involving one site to extensive multiorgan vasculitis leading to death [141, 142].
Epidemiology
GPA most commonly affects individuals in the fourth or fifth decades of life [141, 143]. The disease can, however, affect people at any age (age range, 5–91 years), with approximately 15 % of cases beginning before 20 years of age [144, 145]. There is no gender predilection; it is most common in Caucasians (97 %) and is quite rare in African Americans (2 %) [141, 146].
Pathogenesis
GPA is considered part of a group of autoimmune disorders known as antineutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis, which also includes microscopic polyangiitis, Churg–Strauss syndrome, and renal-limited vasculitis [142, 147]. These disorders are characterized by the presence of necrotizing small vessel vasculitis with the presence of serum autoantibodies directed against neutrophil cytoplasmic constituents, in particular, proteinase 3 (PR3) and myeloperoxidase (MPO). ANCA tests can be performed by both indirect immunofluorescence assay and antigen-specific enzyme-linked immunosorbent assay (ELISA) [148, 149]. ANCA are believed to play a role in the pathogenesis of vasculitis via activation of neutrophils, but most importantly, they are useful in the diagnosis and management of these disorders. The specificity of cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCA) in biopsy-proven disease has been shown to be 90 %. The sensitivity depends on the extent and the activity of the disease. c-ANCA is not detected in most patients at complete remission; rising levels may be indicative of relapse [150].
The most commonly postulated scenario for ANCA-mediated vasculitis is the interaction of neutrophils and the endothelial cells via cell adhesion molecules, which results in releasing cytokines with damage to the vascular walls [151, 152]. Although many advances have occurred in the understanding of the pathogenesis of GPA, so far, it is still unknown how tolerance to the autoantigen PR3 is broken and how the immune response is sustained. PR3 induces dendritic cell maturation via the protease-activated receptor (PAR)-2 and evokes a strong Th1-type T-cell response in GPA [153]. Clusters of PR3-positive cells (neutrophils and monocytes) surrounded by antigen-presenting cells (Th1-type, CD4+CD28 effector memory T cells) and maturing B and plasma cells are found in the granulomatous responses of GPA lesions [154]. Several important studies have characterized the role of PR3 and PAR-2 in the initiation of this process. These might provide evidences for a more target-specific anti-inflammatory therapy for these patients.
Clinical Presentation
Ocular symptoms may be the first manifestation of GPA in 16 % of patients [155]. Orbital involvement is the most common manifestation, and it has been reported in 45–70 % of patients. The orbital inflammation can cause pain, diplopia, proptosis, and bone erosion and can lead to vision loss. Other ocular manifestations of GPA are peripheral ulcerative keratitis, scleritis, and, less commonly, uveitis, retinal vasculitis, and optic neuropathy [156, 157].
Histopathology
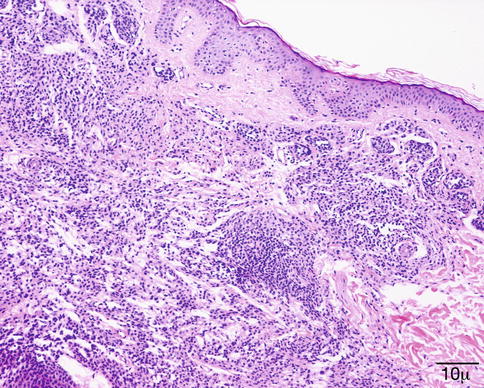
The histological diagnosis is based on the presence of necrobiotic coagulative necrosis, a large number of neutrophils and eosinophils forming small clusters and intermixed with the necrosis; it is associated with histiocytic granulomatous reaction around the large areas of necrosis and small vessel vasculitis. The full picture of necrotizing vasculitis with granulomatosis is seen in the skin in a minority of cases (Fig. 11.31). Sometimes, the findings are quite nonspecific with only chronic inflammation present in the dermis.
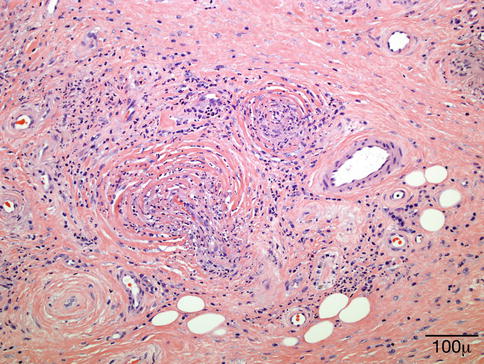
Fig. 11.31
Granulomatosis with polyangiitis. This high power shows the presence of small-vessel vasculitis (hematoxylin-eosin stain; original magnification 200×)
Prognosis
The majority of the patients respond well to treatment, although 25–40 % of patients will experience flare-ups. Anatomical problems are common, such as tracheal stenosis, and may require surgery. Long-term complications are common.
11.7 Benign Lesions
11.7.1 Epidermal Inclusion Cyst
Definition
A cystic invagination of the surface squamous epithelium, which has lost continuity with the surface epithelium
Synonym
Epidermoid inclusion cyst
Epidemiology
It can be congenital or secondary to trauma or surgery. Milium is a small retention cyst caused by obstruction of the orifices of the pilosebaceous units and rarely has clinical significance. The larger lesions are more clinically evident and can occur in the ocular region.
Clinical Features
Epidermal inclusion cyst is a slowly enlarging, firm, globular lesion involving the dermis or subcutaneous tissue (Fig. 11.32). It is a mobile, painless lesion covered by intact epidermis. It can occur as a very small lesion (milium) or as a large lesion.
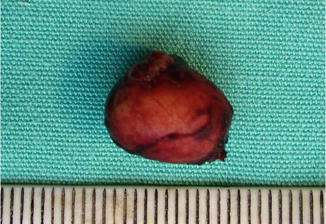
Fig. 11.32
Epidermoid cyst – macroscopic photograph of an intact cyst
A milium appears as one or more small, gray-white, sometimes umbilicated lesions ranging from 1 to 3 mm in diameter. It often has a small keratin plug (blackhead) near the center.
Macroscopy
Thin-walled cyst containing soft material
Histopathology
Epidermal inclusion cysts are lined by keratinizing epithelium and contain keratin (Fig. 11.33). Unlike a dermoid cyst, the lining does not contain dermal appendages. A ruptured epidermoid cyst can incite a severe granulomatous reaction (keratin granuloma) and reactive hyperplasia of the overlying epithelium (Fig. 11.34).
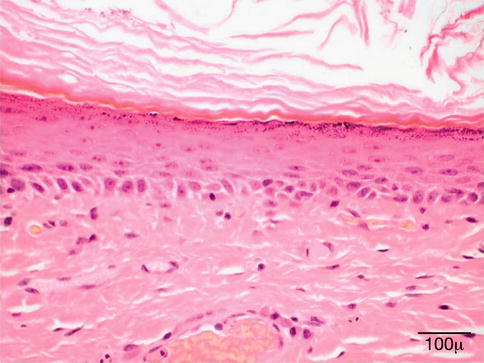
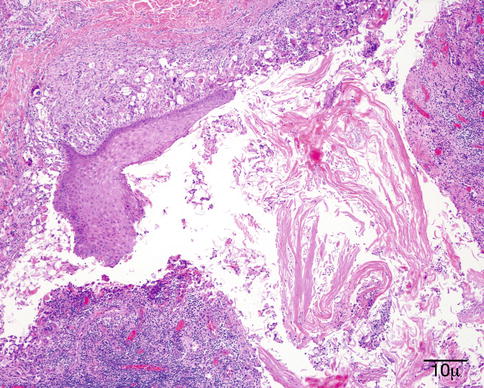
Fig. 11.33
Epidermoid cyst – Cyst lined by organized keratinized squamous epithelium filled with keratin in lumen (hematoxylin and eosin stain; original magnification 200×)
Fig. 11.34
Epidermoid cyst – Ruptured cyst with organizing inflammation in cyst wall and adjacent tissue (hematoxylin and eosin stain; original magnification 100×)
Prognosis
Epidermal inclusion cysts are best managed by local complete excision. An eyelid crease incision provides good results, and the prognosis is excellent. Malignant transformation into basal cell carcinoma or squamous cell carcinoma is a very rare event [162].
11.7.2 Hidrocystoma
Definition
Hidrocystoma is a cystic form of sweat gland adenoma resulting from proliferation of the apocrine or eccrine secretory glands.
Etiology
An obstruction of the sweat gland ducts immediately above the glandular coil within the deep dermal layer following an inflammatory process or trauma [163]. Hidrocystomas are benign lesions of the eyelid that are differentiated into two histological types: apocrine and eccrine.
The apocrine hidrocystoma or cyst of Moll affects the eyelid border and generally appears following an obstruction of the apocrine secretory duct of the gland of Moll.
The eccrine hidrocystoma or cyst of the eccrine sweat gland originates from the eccrine sweat gland, also known as the gland of Moll. These lesions are regarded as ductal retention cysts and have a similar clinical appearance to that of apocrine hidrocystoma [1]. Eccrine hidrocystomas develop from retention of sweat. Heat, humidity, and perspiration seem to cause these lesions to become larger, more numerous, and symptomatic. Hence, these tend to increase in number and size in summer and decrease in the winter months [162].
Clinical Features
The most common site for apocrine cysts of Moll is close to the eyelashes, the route of lacrimal drainage. Apocrine hidrocystoma is almost always a solitary lesion. It consists of small, painless, round, translucent, fluid-filled vesicles (Fig. 11.35) [163] and frequently has a bluish color and may resemble a blue nevus or melanoma [163]. It can range from 1 mm to more than 1 cm in diameter and is filled with clear or milky fluid [163].
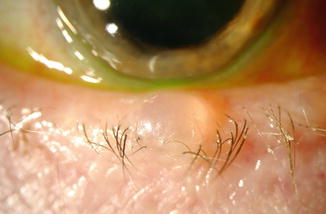
Fig. 11.35
Cyst of Moll (apocrine cyst) – Clinical photograph of well-circumscribed, thin-walled cyst on lower lid
Eccrine hidrocystomas generally present as multiple cutaneous vesicles on the lower eyelid [163], in contrast to apocrine hidrocystoma. Eccrine hidrocystomas develop from retention of sweat.
Histopathology
The apocrine hidrocystoma is a cystic lesion with a clear lumen lined by a double layer of cells (Fig. 11.36). The innermost cells are columnar and display an eosinophilic cytoplasm with atypical bulbous expansions typical of cells undergoing decapitation secretion (Fig. 11.37). The outermost layer consists of flattened myoepithelial cells [162]. The apocrine hidrocystoma may be regarded as a form of papillary cystadenoma rather than as a retention cyst [1].
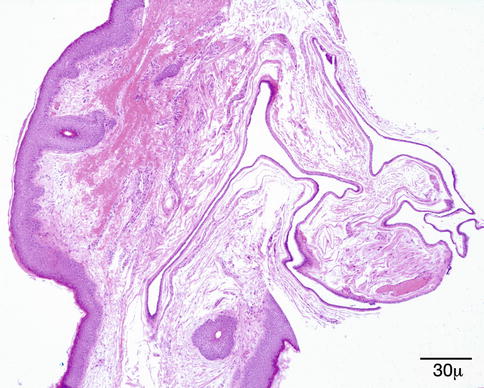
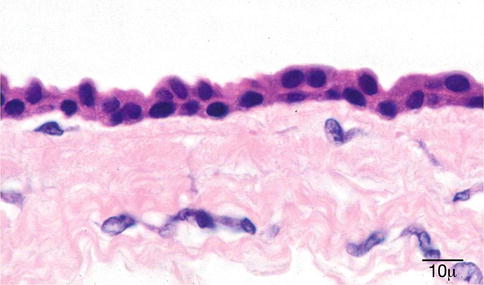
Fig. 11.36
Cyst of Moll (apocrine cyst) – Multiloculated cyst lined by low cuboidal epithelium (hematoxylin and eosin stain; original magnification 40×)
Fig. 11.37
Cyst of Moll (apocrine cyst) – Shows cuboidal epithelium with abundant eosinophilic cytoplasm, which has a granular appearance (hematoxylin and eosin stain; original magnification 100×)
The eccrine hidrocystoma contains only one cystic cavity, which is usually partially collapsed and contains no papillary infoldings. The cyst is usually lined by two layers of small cuboidal epithelial cells; occasionally, only one layer is present. No myoepithelial cells are observed [162].
Prognosis
Prognosis of both forms of hidrocystoma is excellent.
11.7.3 Pilar Cyst
Synonym
Sebaceous cyst
Definition
A pilar cyst is usually secondary to occlusion of the duct of a sebaceous gland and can occur on the eyelid and adjacent tissue.
Etiology
It most commonly develops in the meibomian glands of the upper tarsus from retention of meibomian gland material. Less often, it arises from the Zeis glands near the eyelid margin.
Localization
Larger pilar cysts tend to arise in areas where there are numerous large hair follicles. It commonly develops on the scalp (90 %), occasionally in the eyebrow area, and less often in the medial canthus and eyelid [164].
Clinical Features
Pilar cysts of the meibomian gland are a subcutaneous nodule. The extra-meibomian sebaceous cyst is a slow-growing, freely mobile, smooth, subcutaneous lesion, most often in the region of the eyebrow. It often contains a waxy comedo in the center. It is usually solitary but can be multiple [162].
Histopathology
Pilar cysts are characterized by an epithelial lining that does not possess intracellular bridges. The epithelial cells are not stratified as is seen in normal epithelium. The cells on the inner aspect are enucleate (Fig. 11.38). The lumen of the cyst usually contains homogeneous, eosinophilic material. Calcification occurs in about 25 % of cases. The cyst can also rupture and incite a foreign body giant cell reaction [162].
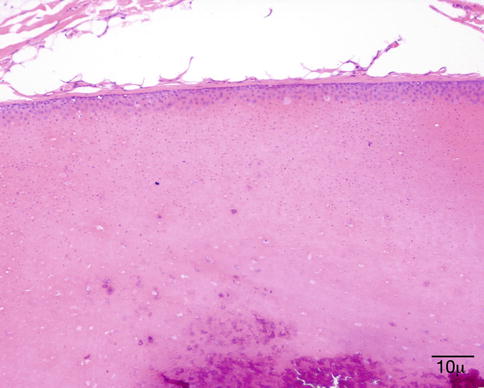
Fig. 11.38
Pilar cyst – Cyst wall showing that the epithelial cells “drift” into the lumen (hematoxylin and eosin stain; original magnification 100×)
Prognosis
A small, asymptomatic pilar cyst can be managed by observation or compress. It eventually resolves in most cases. A larger or symptomatic lesion can be managed by surgical excision.
11.7.4 Dermoid Cyst
Synonyms
Benign cystic teratoma
Definition
Dermoid cyst is a congenital cystic lesion that can affect the eyelid and/or the orbit.
Etiology
Entrapped ectoderm at a site of embryologic bony fusion
Localization
Most dermoid cysts in the ocular region occur at the bony orbital rim superotemporally at the site of the zygomaticofrontal suture as a swelling beneath the lateral aspect of the upper eyelid and eyebrow [162].
Clinical Features
A dermoid cyst is usually slow growing and appears as a smooth, oval-shaped, subcutaneous mass that is not movable because of its attachment to the underlying periosteum (Fig. 11.39) [165]. It usually is located deep in the eyelid skin superotemporally and can occasionally be associated with a fistula that opens through the skin into the eyelid [162]. It can measure from 1 to several centimeters in diameter.
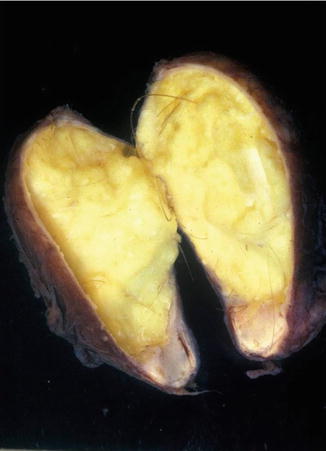
Fig. 11.39
Dermoid cyst – Macroscopic photograph of opened cyst showing cheesy material and hair in wall of cyst
Macroscopy
Thin-walled cyst containing soft, flocculent yellow-white material
Histopathology
Dermoid cysts are lined by stratified epithelium that usually produces keratin, which accumulates in the lumen. The walls of the cyst typically possess adnexal structures, particularly hair follicles, sebaceous glands, and eccrine sweat glands (Fig. 11.40).
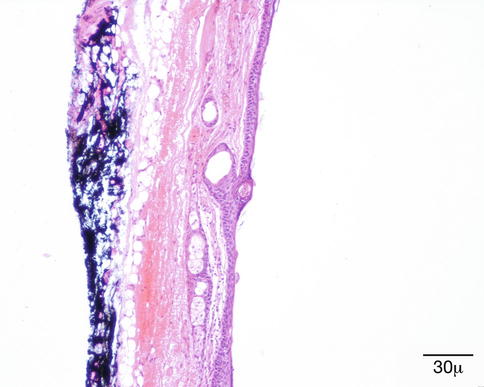
Fig. 11.40
Dermoid cyst – Cyst wall showing skin appendage structures in wall of cyst. This distinguishes a dermoid cyst from epidermoid cyst
Differential Diagnosis
Epidermoid or pilar cyst
Prognosis
The treatment consists of complete extirpation. It is important to maintain the integrity of the cyst during surgery since a loss of internal cyst material or remains will usually provoke an intense foreign body granulomatous reaction in the orbit or eyelid [165].
11.7.5 Seborrheic Keratosis
Definition
Seborrheic keratosis (SK) is a common benign tumor of the epidermis that appears in early adulthood and increases in frequency with age.
Synonyms
Senile wart, seborrheic verruca
Epidemiology
SK is more common in whites than in dark-skinned people. There is no sex predilection.
Etiology
The etiology of seborrheic keratosis is unclear. Aging, cumulative UV radiation-induced mutations, and HPVs are proposed as etiologic factors. However, the role of HPVs is contestable, as the prevalence of HPV positivity differs on top of different skin lesions from the prevalence inside the same tumor; 79 % of SKs were HPV positive on the surface but only 19 % inside the tumor tissue [166].
Localization
SK occurs anywhere on the skin surface, with the exception of the palms and soles. They are quite common in the periorbital area.
Clinical Features
SKs present clinically as sharply demarcated pink, tan, or brown papules or plaques with a verrucous surface. Multiple SKs may appear over a short time as a paraneoplastic phenomenon (Leser–Trélat sign). Irritated SKs are surrounded by an erythematous halo and may mimic BCC or squamous cell carcinoma (SCC).
Macroscopy
The size of the lesions varies from a few millimeters to centimeters. The transition between SK and the surrounding normal skin is usually sharply defined.
Histopathology/Immunoprofile
Multiple variants of SK exist. The lesion may be flat, endophytic, or exophytic. The surface is papillomatous; the epidermis is hyperkeratotic and parakeratotic (Fig. 11.41).
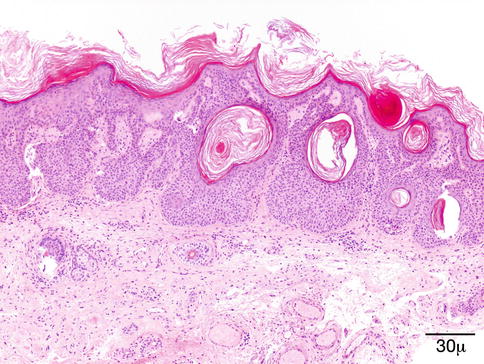
Fig. 11.41
Seborrheic keratosis – Papillomatous projections of digitated SK may mimic a verruca. Broad anastomosing rete ridges and lack of viral cytopathic changes help the differential diagnosis (hematoxylin-eosin stain; original magnification 40×)
The acanthotic type shows a thickened epidermis that extends upward and is composed of cytologically uniform, small basaloid, cuboidal keratinocytes. Horny invaginations and pseudo-horn cysts are apparent.
In the reticulated (adenoid) type, a delicate network of interweaving epithelial strands extends into the dermis. Hyperpigmentation of the basaloid cells may be prominent (Fig. 11.42). The features are reminiscent of lentigo senilis.
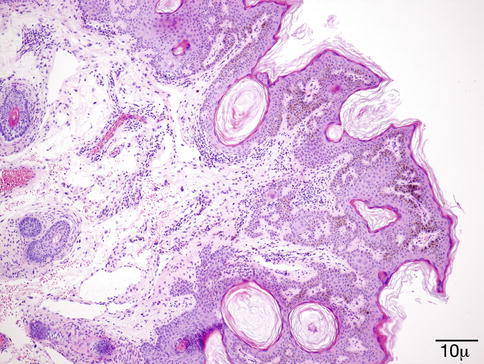
Fig. 11.42
Seborrheic keratosis – This SK exhibits a papillomatous and tentatively reticulate growth pattern with basilar pigmentation. The latter is also a common feature (hematoxylin-eosin stain; original magnification 100×)
Irritated SKs display a more pronounced inflammation and numerous squamous eddies. The proliferating keratinocytes may be spindled and proliferate downward; thus, the horizontal demarcation usually seen in non-irritated SKs is broken.
In the clonal type of SK, well-defined nests of cells with slightly enlarged nuclei are located within the epidermis.
Further variants are the hyperkeratotic, digitated, stucco, and inverted follicular keratosis. The latter demonstrates a marked endophytic growth, but the pattern is lobular and noninfiltrative. Squamous eddies are a prominent feature.
Differential Diagnosis
Epidermal nevus, actinic keratosis, SCC in situ, verruca vulgaris, keratotic melanocytic lesion, squamous papilloma, and acanthosis nigricans
Histogenesis
Proliferating keratinocytes are considered the cells of origin.
Genetics
SK is clonal in nature [167]; but no chromosomal imbalance (a common finding in both benign and malignant tumors) was demonstrated by comparative genomic hybridization in a limited number of SKs [168]. Recently, activating mutations in the fibroblast growth factor receptor 3 (FGFR3) gene have been reported to cause benign skin tumors [169]. UV light-induced somatic mutations of FGFR3 occur in a significant proportion of adenoid-type SKs [170]. In another study of multiple SK samples from four patients, 59 % of the tumors harbored FGFR3 mutations, at least, in four loci in each individual patient [171].
Prognosis and Predictive Factors
SKs are benign lesions, although they are a cosmetic concern. However, they may be associated with Bowen disease or rarely BCC. Since they also may mimic malignant tumors, they are biopsied to provide a histopathological diagnosis.
11.7.6 Inverted Follicular Keratosis
Definition
A benign, warty tumor of the epidermis that increases in frequency with age
Epidemiology
Occurs mainly in middle-aged to older adult men, more commonly in Caucasians
Etiology
It may develop rather rapidly over a few months and is believed to have a viral etiology. It was once believed to be of a hair follicle origin. However, there has been no convincing histopathological evidence to support that concept and is now believed to be an inverted irritated seborrheic keratosis [162].
Localization
It is often located on or very near the eyelid margin.
Clinical Features
It has a wartlike appearance that may be pigmented, stimulating a melanocytic lesion.
Histopathology
The epithelium exhibits downward lobular hyperplasia with proliferation of both squamous and basal cells with an endophytic pattern and smooth interface with underlying dermis (Fig. 11.43). Squamous eddies are frequently observed.
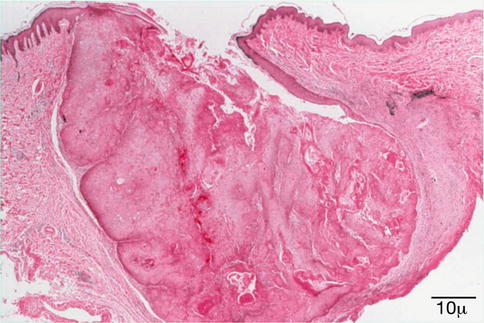
Fig. 11.43
Inverted follicular keratosis – Downward hyperplasia of epithelium with smooth interface between epithelial basement membrane and dermis (hematoxylin-eosin stain; original magnification 100×)
Differential Diagnosis
Any pigmented or nonpigmented keratotic lesion, including seborrheic keratosis, actinic keratosis, pigmented melanocytic lesions, and basal cell carcinoma
Prognosis
Benign course; shave excision is usually sufficient.
11.8 Premalignant Conditions
11.8.1 Actinic Keratosis
Definition
Actinic keratosis (AK) is a biologically benign, noninvasive epidermal dysplasia developing in sun- or other radiation-damaged skin.
Synonyms
Solar keratosis
Epidemiology
AK affects mostly fair-skinned, elderly patients with a history of chronic UV exposure from sunlight. AKs are more frequent in men with skin types I and II; 73 % of patients with SCC have AKs as well, according to a case-control study in the Netherlands [172]. Furthermore, the same study disclosed that painful sunburns before the third decade of life were also associated with SK.
Etiology
Both environmental and genetic risk factors play a role in the development of AK. UVB-induced mutations of the p53 gene, mutations in ras genes, c-Myc proto-oncogenes, and mutations in tumor suppressor genes, such as p16INK4a, have all been implicated in the pathogenesis of AKs.
Localization
Sun-exposed areas (face, scalp, ears, dorsum of the hands), common in the periorbital region, but rare on the eyelids
Clinical Features
Presents as small, scaly, irregular-shaped, erythematous plaques or papules
Histopathology/Immunoprofile
Although several variants of AK are recognized, all lesions are characterized by atypia of the basal cell layer of the epidermis with elongated hyperchromatic nuclei; eventually, focal suprabasilar atypia is also present (Fig. 11.44). The epidermis is parakeratotic, and the dermis exhibits solar elastosis and dilated vessels. The epidermis is usually thickened but may be atrophic. The histological variants include hypertrophic, acantholytic, proliferative, pigmented, and lichenoid [173].
Fig. 11.44
Actinic keratosis – The epidermis is thickened, suprabasilar keratinocytes show cytoplasmic pallor (hematoxylin-eosin stain; original magnification 600×)
Differential Diagnosis
Differential diagnosis includes squamous cell carcinoma and seborrheic keratosis. Pigmented AK should be differentiated from solar lentigo and lentiginous melanoma in situ. It is worth mentioning here that melanoma is extremely rare on the eyelids.
Genetics
Chromosomal alterations and genomic instability [175] are present in AK, leading to increased signals that facilitate proliferation (Ras, Bcl-2) and decreased signaling of p53 as well as molecules regulating keratinocyte proliferation and differentiation [176]. Microarray analysis of AKs, SCCs, and the normal skin from 16 patients revealed 89 genes similarly affected in AK and SCC [177]. Genes that were upregulated in AK and SCC were downregulated in the normal skin and vice versa.
Prognosis and Predictive Factors
AK can regress spontaneously. There is disagreement about the rate of progression from AK to invasive SCC, but 60–97 % of SCCs supposedly originate from AKs [178].
11.8.2 Bowen Disease (Squamous Cell Carcinoma In Situ)
Definition
Bowen disease (BD) is a form of carcinoma in situ of the epidermis.
Epidemiology
It affects mostly individuals in their sixth to eighth decade; men and women are equally affected. There is no valid estimate of the frequency in the eyelids.
Etiology
UV radiation, radiation therapy, and various types of HPV also may have a role, namely, HPVs 16, 18, 2, 31, and 33. Arsenic exposure [179] that causes the production of ROS [132] from medications, contaminated well water, and pesticides have also been associated with BD. Immunosuppressed patients are increasingly at risk [180].
Localization
BD has a predilection for the sun-exposed areas of the body. In males, it affects mostly the head and neck region, while in females, it localizes predominantly to the lower limbs and cheeks.
Clinical Features
It presents as a well-defined, scaly erythematous plaque. Nodular, verrucous, eroded, and pigmented variants are observed.
Histopathology/Immunoprofile
The epidermis is hyperkeratotic and acanthotic. Atypical keratinocytes involving the full thickness of the epidermis are associated with loss of maturation in the stratum spinosum as well as loss of the granular layer (Fig. 11.45). However, the presence of an intact layer of basal cells [181] is frequently observed, at least, in early lesions [174]. Pagetoid spread of large, pale tumor cells with ground-glass cytoplasm is common. Adnexal structures are frequently involved (Fig. 11.46). Several histological variants exist, and more than one pattern may be present in different areas of the same lesion. Chronic, moderate inflammation in the underlying dermis is common. The different variants include atrophic, verrucous-hyperkeratotic, irregular, and pigmented. BD is positive for AE1–AE3 and CK 10 and does not stain for CAM5.2 (CK18). Clear cell BD express CK13, CK16, and CK15, which are cytokeratins found in the outer root sheath. MIB-1 antibody marks the suprabasal layers more extensively than the basal layer of columnar cells [174].
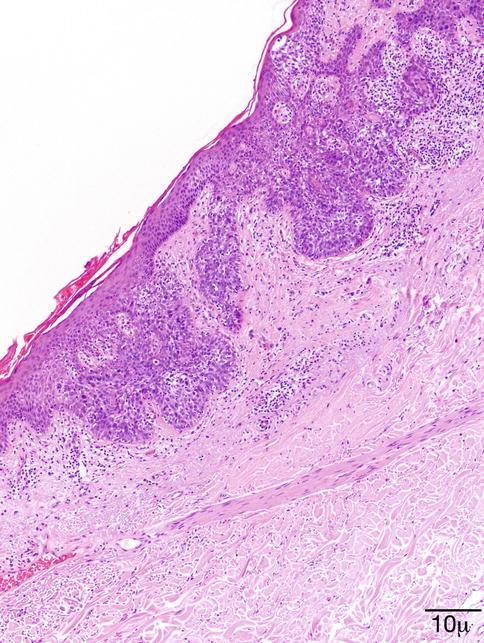
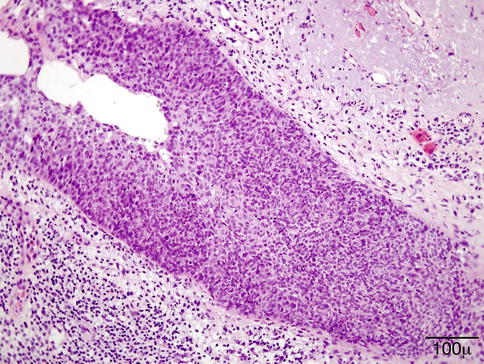
Fig. 11.45
Bowen disease – Loss of polarity and cellular atypia mostly localized to spinous cell layer is characteristic of BD. Cytopathic changes related to HPV are also present in this case (hematoxylin-eosin stain; original magnification 100×)
Fig. 11.46
Bowen disease – Intraepithelial extension of BD into adnexal structure should not be misinterpreted as stromal invasion (hematoxylin-eosin stain; original magnification 200×)
Differential Diagnosis
Actinic keratosis, squamous cell carcinoma, Paget disease, superficial BCC
Genetics
Prognosis and Predictive Factors
The prognosis is fair; only about 3–5 % of cases progress to invasive squamous cell carcinoma.
11.9 Neoplasms
11.9.1 Epithelial Neoplasms
11.9.1.1 Squamous Papilloma
Definition
Squamous papilloma (SP) is a benign tumor of epithelial origin.
Synonyms
Fibroepithelial polyp, skin tag, acrochordon
Epidemiology
Squamous papilloma is the most common benign tumor of the eyelid. Most occur between the age of 30 and 50 years.
Etiology
Various types of low-risk HPVs are implicated in the development of squamous papilloma.
Localization
Squamous papilloma has a predilection for the eyelid border; multiple lesions may be present.
Clinical Features
Squamous papillomas are slow-growing, sessile or pedunculated pointy outgrowths. The color depends on the amount of surface keratin. Heavily keratinized lesions are whitish, while lesions with less keratin are skin colored.
Histopathology
Squamous papilloma exhibits fingerlike projections of fibrovascular core covered by hyperplastic epidermis with elongated rete ridges and acanthosis. Focal parakeratosis is present (Fig. 11.47).
Fig. 11.47
Squamous papilloma – Branching fibrovascular core covered by a variably hyperplastic, hyperkeratosic squamous epithelium (hematoxylin-eosin stain; original magnification 40×)
Differential Diagnosis
At scanning, magnification of the silhouette of verruca vulgaris and seborrheic keratosis may be similar. Verruca may be distinguished by koilocytosis, while seborrheic keratosis may have horn pseudocysts absent in squamous papilloma.
Histogenesis
Squamous papilloma may originate from the surface epithelium and from mucous membranes.
Prognosis and Predictive Factors
The prognosis of eyelid papillomas is excellent. Malignant transformation does not occur. However, treatment may be needed for cosmetically disturbing lesions. A shave excision is usually sufficient.
11.9.1.2 Basal Cell Carcinoma and Variants
Definition
Basal cell carcinoma (BCC) is the most common skin cancer among Caucasians. It occurs mainly on the sun-exposed areas of the body (face, scalp, periocular area, upper extremities).
Synonyms
Basalioma, basal cell epithelioma
Epidemiology
BCC is the most common malignant tumor of the eyelid skin [184] and accounts for 90 % of all malignant tumors of the eyelids. It affects mainly fair-skinned people over 40 years of age. The male-to-female ratio is between 2:1 and 1.6:1.
Etiology
UV light is considered the principal etiologic factor in both sporadic BCC and BCC associated with a genetic defect (Gorlin syndrome, albinism, Bazex–Dupré–Christol syndrome, xeroderma pigmentosum). Other factors are ionizing radiation and arsenic exposure. Indoor tanning is a culprit recently recognized [185]. Patients affected by BCC are at an increased risk of developing new skin cancers [186, 187].
Localization
BCC is unevenly distributed around the eye. The localization of BCC in order of frequency is the lower eyelid, medial canthus region, upper eyelid, and lateral canthus region. Interestingly, the distribution of periocular BCCs is unrelated to the amount of solar UV radiation to the periocular area [188], the former being evenly scattered over the lids and canthi. BCC tends to involve the eyelid margin with loss of lashes (madarosis).
Clinical Features
BCCs are slowly growing, locally invasive tumors that rarely metastasize but can cause extensive tissue destruction. The most common clinical presentation is a painless pearly nodule with rolled borders and surface telangiectasia (Fig. 11.48). The tumor may exulcerate and bleed, with central crusting (Fig. 11.49). Cystic tumors are translucent with a bluish tinge. The infiltrating type has ill-defined borders and presents as a pink-to-tan plaque. The surface may be depressed (Fig. 11.50). The lesion is firm to touch. Pigmented BCC may be mistaken for malignant melanoma.
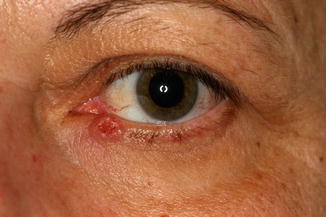
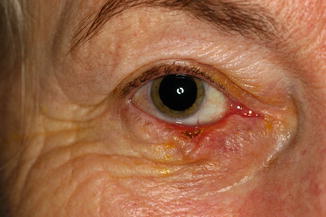
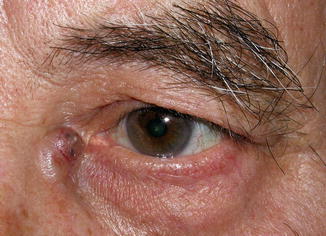
Fig. 11.48
BCC – Small nodular tumor of the lower eyelid. Pearly ulcerated nodule with rolled up edges.
Fig. 11.49
BCC – Ulcerated tumor of the lower lid of a middle-aged woman. The tumor’s border is ill-defined, notice the loss of lashes (madarosis)
Fig. 11.50
BCC – Irregularly pigmented BCC at the nasal canthus of an elderly man
Macroscopy
The most common specimen is an oval or pentagonal (when the tumor is close to or involves the eyelid margin) excision with a well-circumscribed, sometimes cystic, nodular tumor that is either ulcerated or not ulcerated. The sclerosing variant diffusely infiltrates the surgical specimen with whitish strands of tumor tissue blending into the surroundings. An exenteration specimen may eventually be received for a BCC with extensive orbital spread.
Histopathology/Immunoprofile
BCCs are frequently of mixed growth pattern. One pattern may predominate or be present in a pure form.
Nodular BCC is the most common type. It is composed of lobules of basaloid cells with a pushing border (Fig. 11.51). Peripheral palisading and peritumoral clefting with presence of a myxoid stroma are usually present. The tumor cells have a high nuclear-to-cytoplasmic ratio; condensed, finely granular chromatin; inconspicuous nucleoli; and a variable mitotic activity. Apoptotic cells are numerous. An inflammatory reaction in the stroma is composed of a variable population of lymphocytes, plasma cells, and histiocytes.
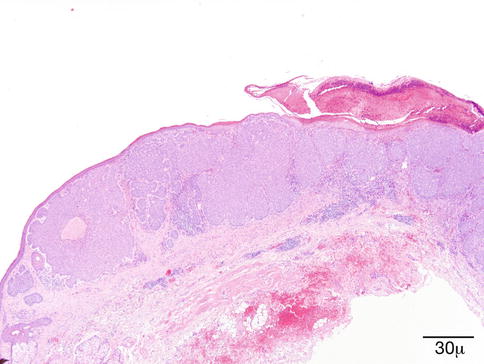
Fig. 11.51
BCC – Nodular aggregates of basaloid cells infiltrate the dermis. Peripheral palisading and clefts between tumor cells and surrounding stroma are conspicuous (hematoxylin-eosin stain; original magnification 40×)
Superficial BCC is characterized by multifocal aggregates of basaloid tumor cells with multiple connections to the epidermis, separated from each other by areas of the normal skin. Extensive regression may occur and accentuate the seemingly multifocal pattern.
Infiltrative and morpheaform BCC exhibits narrow cords and strands of cells, often accompanied by a prominent stromal reaction and increased collagen deposition. Small, irregularly shaped nodules with only focal or absent peripheral palisading are observed. Stromal mucin and retraction artifact are also missing (Fig. 11.52).
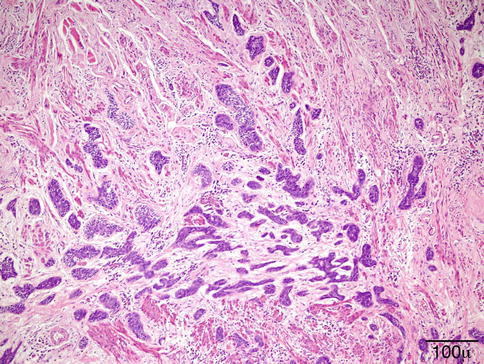
Fig. 11.52
BCC – Infiltrative pattern is characterized by small, irregularly shaped groups or narrow pointed strands of tumor cells. Recurrence rate is high (hematoxylin-eosin stain; original magnification 200×)
Adenoid BCC usually has a nodular growth pattern with increased stromal mucin filling cribriform or pseudoglandular spaces (Fig. 11.53).
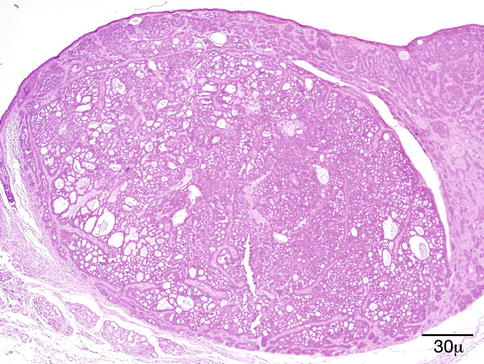
Fig. 11.53
BCC – Tumor with adenoidal features. The nodular tumor shows a cribriform pattern (hematoxylin-eosin stain; original magnification 40×)
Pigmented BCC is most often of the nodular type. Only about 1 % of BCCs are pigmented. Melanin pigment is found in melanophages, tumor cells, or both [189]. BCC may collide with a melanocytic proliferation.
BCC with pseudoepitheliomatous hyperplasia is probably due to ulceration or irritation by scratching. Immunohistochemical studies may be necessary to differentiate these cases from SCC. Strong Ber-EP4 positivity and weak focal EMA favor BCC [190, 191].
BCC with squamous differentiation in the deeper infiltrative part of the tumor is seen in 1–3 % of BCCs, mostly in recurrent tumors (Fig. 11.54).
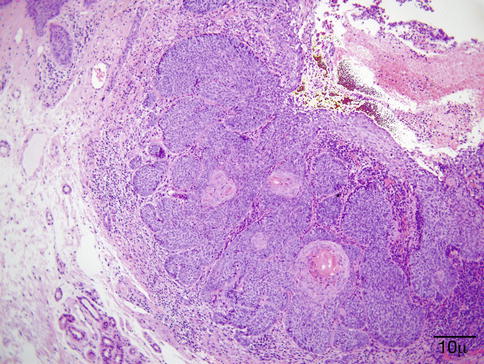
Fig. 11.54
BCC – Squamous differentiation occurs most frequently in ulcerated BCCs, as seen in this otherwise nodular tumor. Dyskeratotic cells are located within the nodules (hematoxylin-eosin stain; original magnification 100×)
Metatypical and basosquamous BCC can be defined as a BCC differentiating to SCC with intermediate transition zone linking the two [190, 191].
Pleomorphic BCC has marked cellular pleomorphism with huge nuclei but also displays the usual features of conventional BCC.
All variants of BCC are strongly Ber-EP4 positive. The basal layer is CD10 positive [192]. They are negative with CK7, AE1–AE3, CAM5.2, and CK20.
Differential Diagnosis
A number of basaloid tumors may mimic BCC. Desmoplastic trichoepithelioma is difficult to differentiate from infiltrative/morpheaform BCC. The presence of Merkel cells in trichoepithelioma can be highlighted by CK20. Merkel cells are not seen in morpheaform BCC [192]. Mitotic activity and severe solar elastosis favor BCC.
Basal cell carcinoma with squamous metaplasia may be difficult to distinguish from squamous cell carcinoma. If areas typical of BCC are absent, Ber-EP4-positive and EMA-negative staining is helpful.
Morpheaform BCC may be confused with the basaloid proliferation of microcystic adnexal carcinoma. Cytological atypia is more pronounced in BCC; keratocysts are absent.
Merkel cell carcinoma (MCC) may clinically and histologically mimic BCC. The distinction is extremely important, owing to the aggressive behavior of MCC, its propensity to metastasize early in the process of the disease. The nuclei in MCC have a finely granular chromatin and brisk mitotic activity, and apoptotic bodies are conspicuous. CK20 shows a dot-like perinuclear pattern in MCC.
Histogenesis
It is generally accepted that BCC arises from epidermal stem cells located in the bulge of the hair follicles. It is unassociated with a precursor lesion [193].
Genetics
The hedgehog signaling pathway is deregulated in BCC. The gene most often altered in sporadic BCCs is the PTCH gene. Loss-of-function mutations in patched 1 (PTCH1) happen in one-third of sporadic BCCs [194]. Under normal conditions, PTCH inhibits Smoothened (Smo), which functions as a signaling molecule for the expression of downstream target genes involved in transcription and proliferation [195, 196]. UV-specific nucleotide changes (C→T; CC→TT) are detected in both the TP53 and PTCH tumor suppressor genes [197, 198]. Some sporadic BCCs harbor mutations in both alleles of the PTCH gene; however, the mutations are not always characteristic of UV-induced mutagenesis, even in tumors of the sun-exposed skin [199]. Point mutations in the p53 tumor suppressor gene and mutations in the oncogene CDKN2A, a cell cycle regulator, also have a high frequency in BCC.
Prognosis and Predictive Factors
Basal carcinoma of the eyelids is traditionally treated by surgery with margin control. The prognosis for primary nodular tumors is excellent if the excision is complete. In a study involving 270 patients with periocular BCC who underwent 413 procedures, none of the patients with primary nodular BCC suffered recurrence during a 5-year follow-up period [186].
Incomplete primary excision is recognized as the main risk factor for further recurrences [41, 200]. Incomplete excision is more common with morpheaform tumors and medial canthus location. The medial canthus is associated with a high risk of recurrence and invasion into the orbit and paranasal sinuses. Indeed, infiltrative BCC located at the medial canthus represents the main risk factor for exenteration [200]. Positive margins in exenteration specimens may predict intracranial invasion. These data underscore the necessity of reliable margin evaluation and report by the histopathologist.
11.9.1.3 Nevoid Basal Cell Carcinoma Syndrome
Definition
Nevoid basal cell carcinoma syndrome (NBCCS) is a rare autosomal dominant condition characterized by increased susceptibility to BCC, as well as the development of other tumors such as medulloblastoma, fibroma, and rhabdomyosarcoma [201]. Patients also have developmental defects: palmoplantar pits, odontogenic keratocyst of the jaw, macrocephaly, and rib and spine malformations.
Synonyms
Gorlin syndrome, Gorlin–Goltz syndrome
Epidemiology
Nevoid basal cell carcinoma syndrome represents 1/30,827–1/164,000 population [202]. It is almost equally distributed among men and women.
Etiology
Clinical Features
The diagnosis is made in the presence of either two major and one minor criteria or one major and three minor criteria. Affected individuals have coarse facial features, macrocephaly, and bossing of the forehead. Medulloblastoma – primitive neuroectodermal tumor (PNET) – appears earlier than the sporadic form. Odontogenic keratocysts develop during the first decade and may be symptomless. Multiple BCCs appear between puberty and the age of 30 years. Milia-like lesions occur on the palpebral conjunctiva.
Histopathology
The BCCs in NBCCS are indistinguishable from sporadic BCCs.
Prognosis
The prognostic criteria are the same for sporadic BCC. Patients with Gorlin syndrome have a normal life expectancy.
11.9.1.4 Squamous Cell Carcinoma and Variants
Definition
Squamous cell carcinoma (SCC) is an invasive tumor of epidermal origin that propagates to the dermis. SCC is able to produce locoregional and disseminated metastases and is potentially lethal.
Epidemiology
SCC is the second most common malignancy of the eyelid skin after BCC. It represents 5–10 % of malignant eyelid tumors. In different case series, there is a slight [41] or pronounced [204] male predominance. Most patients are in their sixth or seventh decade. The highest incidence is reported from Australia. Solid organ transplant recipients and patients with malignant lymphoma or immunosuppression are at increased risk of developing aggressive SCCs [205–207].
Etiology
Chronic UV exposure is the most important etiologic factor; radiation damage, immunosuppression, chemical carcinogens, HPV infection, and DNA repair defects are also increased risk factors for developing SCC.
Localization
SCC of the skin is localized to sun-exposed areas. Its distribution in the periorbital area is similar to that of BCC, the lower eyelid and nasal canthal area being most frequently affected [204]. It has a strong tendency to involve the eyelid margin.
Clinical Features
SCC may manifest as an erythematous, scaly papule or plaque-like lesion, with either well- or ill-defined borders. Larger nodular tumors have a tendency to ulcerate and exhibit irregular rolled edges. Patients with SCC typically have clinical signs of actinic damage, Bowen disease, or actinic keratosis.
Macroscopy
Superficial SCC is indistinguishable from any type of carcinoma, either in situ or actinic keratosis. Invasive SCC is ill-defined, invades the dermis, and may be plaque-like, wedge shaped, or craterlike.
Histopathology/Immunoprofile
In invasive SCC, irregular masses of epidermal cells proliferate downward into the dermis. As the eyelid dermis is sparse, the tumor can easily reach the orbicular muscle fibers. The contour of the proliferating tumor is often jagged. Discohesive growth manifests by small aggregates or even single tumor cells. The cells exhibit various degrees of atypia, nuclear hyperchromatism and pleomorphism, and mitotic figures (Fig. 11.55). Based on the degree of differentiation and keratinization, SCC is graded as well, moderately, or poorly differentiated. However, there is no direct correlation between the degree of differentiation of a given SCC and its biological behavior [208]. Keratinization is well developed in differentiated tumors. With the formation of squamous pearls, the cells are mildly atypical and have abundant pink cytoplasm. In less well-differentiated SCC, keratinization is less evident and cells have more atypia. In poorly differentiated tumors, the tumor cell nuclei are markedly atypical or anaplastic; keratinization is minimal or nonexistent (Fig. 11.56). The dermis may show a focal or diffuse inflammatory infiltrate that is lymphoplasmacellular or neutrophilic.
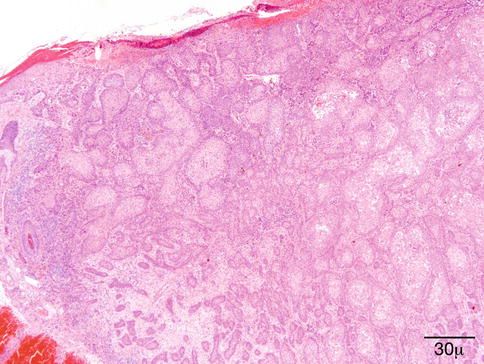
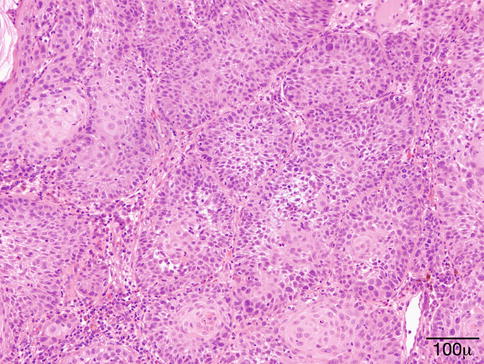
Fig. 11.55
SCC – Well-differentiated invasive SCC from the eyelid of an elderly man. Keratinization is predominant; (hematoxylin-eosin stain; original magnification 40×)
Fig. 11.56
SCC – Poorly differentiated SCC; keratinization is sparse; markedly atypical nuclei are prominent (hematoxylin-eosin stain; original magnification 200×)
SCCs show nuclear positivity for p63. High molecular weight cytokeratin (CK1, CK5, CK10, and CK14) expression is cytoplasmic. Many SCCs are positive for AE1–AE3; however, negativity does not exclude SCC.
Variants
Acantholytic SCC: There is loss of cohesion between the tumor cells; a typical SCC pattern is mixed with pseudoglandular formations.
Clear cell SCC: The cytoplasm of tumor cells is pale due to the presence of glycogen. They should be differentiated from other clear cell tumors such as clear cell carcinoma; sweat gland carcinoma; sebaceous carcinoma (EMA+, AR+, CAM5.2+), the third most common malignant tumor of the eyelids and periocular region; and trichilemmal carcinoma.
Spindle cell SCC: Predominantly composed of spindle cells, if keratinization is absent, confusion with sarcoma, atypical fibroxanthoma, or spindle cell melanoma is possible. Immunohistochemistry is needed to make the differential diagnosis.
Desmoplastic SCC: A prominent desmoplastic reaction in certain tumors with thin cords of epithelial cells (Fig. 11.57) and small keratin cysts may imitate a squamatized morpheaform BCC or a microcystic adnexal carcinoma.
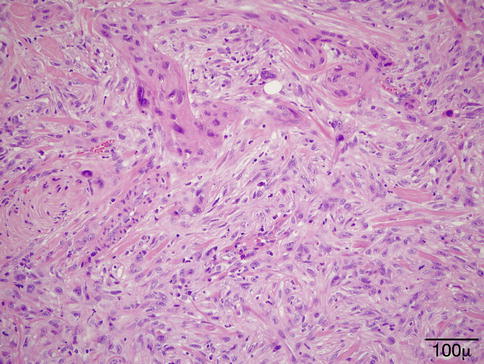
Fig. 11.57
Desmoplastic SCC – At higher magnification, chords of epithelioid tumor cells are recognized (hematoxylin-eosin stain; original magnification 200×)
Differential Diagnosis
Pseudoepitheliomatous hyperplasia (PEH) may resemble SCC. Knowledge of the clinical background (prior surgery, radiation therapy) is important. In PEH, there is no loss of keratinocyte polarity, and the stroma is reactive. Undifferentiated SCCs can mimic melanoma or sarcoma; immunostains for cytokeratins are needed for the correct diagnosis.
Porocarcinoma has the propensity to invade the lymphatics and is lethal in one-third of patients; it has a dismal prognosis compared to SCC [209]. CK15 and carcinoembryonic antigen (CEA) are useful markers in the differential. Porocarcinoma is CK15+ and CEA stains the ducts, while SCC is CK15-, and CEA is positive in tumor cells [210].
Histogenesis
SCC is a tumor of epidermal keratinocytes. It may occur de novo or following the malignant transformation of a precursor lesion, such as actinic keratosis or Bowen disease.
Genetics
The p53 tumor suppressor gene is mutated in 46–72 % of patients with SCC [211]. These mutations are due to direct absorption of UV light by DNA and produce characteristic C→T or CC→TT tandem mutations at dipyrimidine sites [211, 212]. Patients with xeroderma pigmentosum (XP), a rare autosomal recessive disorder, have a defect in UV-induced DNA damage repair due to an inappropriate nucleotide excision repair system. XP patients are predisposed to malignant skin neoplasms in sun-exposed areas. SCC may develop in early childhood. Some patients may have hundreds of skin tumors over time [21].
Loss of heterozygosity (LOH) on chromosome 9q22 has been observed in the tumor tissue in 60.8 % of SCCs, with the highest loss occurring in and around the PTCH gene [213].
Prognosis and Predictive Factors
Excision with clear margins usually cures most SCCs. However, the periorbital region and eyelids are high-risk anatomical sites, and a number of tumors recur even after apparent complete surgical excision. The main risk factor for local recurrence is incomplete primary excision [41]. Perineural invasion and orbital infiltration frequently coexist [204, 214] and seem more common than regional lymph node metastasis, which is relatively low [215]. Even if exenteration is necessary, the prognosis is favorable.
11.9.2 Melanocytic Lesions and Neoplasms
Definition
Melanocytic cells, which by definition contain melanosomes and melanin pigment, normally occur as solitary cells in the dermis and vary in size and number, depending on racial origin. Melanocytes are derived from the neural crest and migrate to the skin during embryogenesis. A neoplastic proliferation of melanocytes may be acquired or congenital.
Benign lesions include ephelis (freckle), benign lentigo, a variety of nevi, and lentigo maligna, which is premalignant, and the malignant melanoma.
The benign melanocytic lesions vary considerably in their clinical appearance. They frequently occur on the surface of the eyelid or on the lid margin.
11.9.2.1 Ephelis
Synonym
Freckle
Definition
A localized increase in melanocytic pigmentation in the basal epidermal cells without a proliferation of melanocytes
Etiology
The histogenesis is hyperactivity of melanocytes, which release melanin into the basal epidermal cells.
Clinical Features
This pigmented lesion appears as a small, brown macule scattered over the sun-exposed areas of the skin, including the eyelids. In contrast to lentigo simplex, sunlight exposure deepens the pigmentation of freckles.
Histopathology
There is hyperpigmentation of the basal cell layer but no elongation of the rete ridges. The basal layer of the epidermis contains large, DOPA-positive melanocytes, but their number is not increased.
Prognosis
Prognosis is excellent. Removal is not warranted unless for cosmesis.
11.9.2.2 Lentigo Simplex
Definition
Lentigo simplex is a benign skin lesion usually observed in younger people. It may occur at any age and is not affected by exposure to sunlight.
Etiology
This includes a localized increase in melanocytes, which are functionally active and secrete melanin. It has been proposed that lentigo simplex may evolve into a lentiginous/junctional melanocytic nevus when melanocytes proliferate and aggregate to form small nests in the junctional zone, whereas solar lentigo may be a precursor lesion of seborrheic keratosis [171].
Clinical Features
The lesions measure approximately 1–2 mm in diameter, are flat and brown to black, and are clinically indistinguishable from a melanocytic nevus and junctional nevi.
Histopathology
Lentigo simplex shows a moderate elongation of the epidermal rete ridges and basal hyperpigmentation (Fig. 11.58). The number of melanocytes in the basal epidermal layer may be slightly increased (Fig. 11.59). In contrast to lentigo simplex, solar lentigo displays markedly elongated rete ridges and profound actinic elastosis in the dermis.
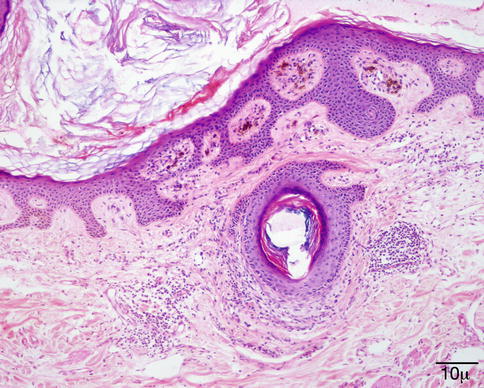
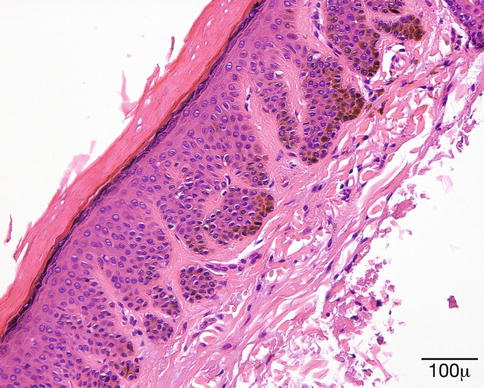
Fig. 11.58
Lentigo simplex – Elongation of rete pegs of epithelium; there is no junctional melanocytic proliferation (hematoxylin-eosin stain; original magnification 100×)
Fig. 11.59
Lentigo simplex – Highlights pigmentation of the basal layer of the epithelium (hematoxylin-eosin stain; original magnification 200×)
Prognosis
The prognosis is excellent. There is no premalignant potential. Multiple lentigines can be found in multiple lentigines syndrome (LEOPARD syndrome), which is associated with electrocardiogram (ECG) conduction defects, ocular hypertelorism and other eye defects, pulmonary stenosis, growth retardation, abnormal genitalia, and sensorineural deafness, and in Carney’s syndrome, which has an autosomal dominant inheritance pattern and is associated with bilateral primary pigmented nodular adrenocortical hyperplasia, cutaneous myxomata, breast lesions, pituitary tumors, and melanotic schwannomas.
11.9.2.3 Solar Lentigo
Solar lentigo is characterized clinically by flat epidermis with microscopic acanthosis and circumscribed pigmentation affecting the sun-exposed skin.
Synonyms
Lentigo senilis, actinic lentigo
Epidemiology
This lesion is mainly seen in middle-aged to elderly Caucasians.
Localization
Solar lentigo is most common in the skin of the face, including the eyelid. This lesion predominantly affects the sun-exposed skin.
Clinical Features
It is characterized by a well-circumscribed macule, flat lesion, with variably brown pigmentation.
Histopathology/Immunoprofile
The epidermal surface is hyperkeratotic. The epidermis shows atrophy of upper layers with elongation of the rete ridges. Rete ridge hyperplasia is less prominent in lesions involving the face. Increased melanin pigmentation is present, especially in the rete pegs. Intraepithelial melanocytes may be of normal number or increased with mild cellular pleomorphism present. Pigment incontinence may be seen in the dermis or in associated macrophages (melanophages). The dermis shows solar elastosis and mild lymphohistiocytic infiltrate.
Differential Diagnosis
It includes pigmented seborrheic keratosis and lentigo maligna.
Prognosis
It is excellent. Uncommonly, lentigo maligna may arise in long-standing solar lentigo.
11.9.2.4 Junctional Nevus
It arises from the deeper layers of the epidermis (“junctional region”) and does not involve the underlying dermis.
Clinical Features
They appear as flat, pigmented lesions.
Histopathology
These appear as nests of melanocytic cells at the dermal–epidermal junction. There is little cellular pleomorphism and no mitotic activity.
Although connected to the epidermis, nevus cells have the capacity of “dropping off” into the dermis, and many nevi that appear to be purely junctional in one area are found to be compound in other areas.
Prognosis
A benign junctional nevus has an excellent prognosis and can be removed locally. This type of nevus, like the compound nevus, has the capacity to evolve into a malignant melanoma. The development of a frank malignant melanoma is associated with increased nuclear pleomorphism, hyperchromatism, increased mitotic activity, and an inflammatory reaction in the underlying dermis.
11.9.2.5 Compound Nevus
This lesion possesses features of both junctional and intradermal nevi (Fig. 11.60). In an appreciable portion of these lesions, nests of intradermal nevus cells show maturation at the dermal base of the lesion. The cells are small and uniform and have little cellular pleomorphism. The amount of intracellular pigment is variable. This type of nevus is more common than the purely junctional nevus and may also undergo malignant changes.
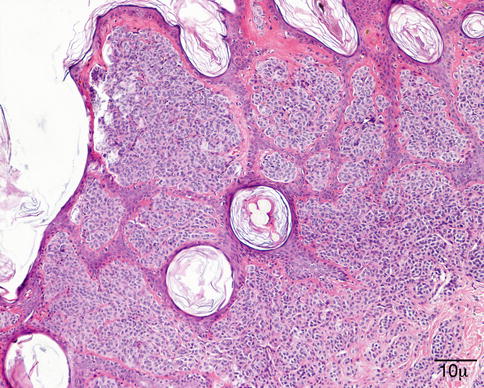
Fig. 11.60
Compound nevus – Proliferation of bland-appearing uniform melanocytic cells in the basal epidermis and dermis. There is no cellular pleomorphism or mitotic activity. Epidermal inclusions are seen. The latter is a benign feature (hematoxylin-eosin stain; original magnification 100×)
11.9.2.6 Dermal Nevus (Intradermal Nevus)
This is the most common of the three types of nevocellular nevi. Malignant change in a purely intradermal nevus is extremely rare.
Clinical Features
It is a pigmented, raised lesion with a well-circumscribed border (Fig. 11.61). The surface of an intradermal nevus may have a papillomatous domed or pedunculated configuration. Clinically, the presence of hairs in an elevated pigmented nodule usually indicates that the lesion is intradermal.
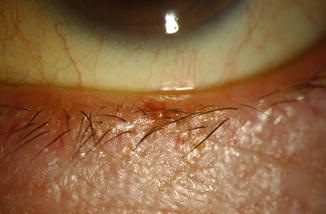
Fig. 11.61
Nevus – Clinical photograph showing a flat pigmented lesion on the lower eyelid
Histopathology
The nests of nevus cells in the dermis are separated from the overlying epidermis by an uninvolved layer of upper dermis (Fig. 11.62). In the eyelid, nevus cells may extend into the deeper dermis and reach the fibers of the orbicularis muscle, giving the false impression of a neoplastic infiltrate. This should not be considered an ominous finding. The presence of nests of large, multinucleated nevus cells may only be found in mature intradermal nevi and are considered to indicate the benign nature of the lesion.