X-Linked Ophthalmic Disorders
Kammi B. Gunton
Irfan Ansari
Leonard B. Nelson
The approach to hereditary ocular disease is undergoing a profound transition with the information provided by modern molecular genetics. The Human Genome Project has begun to provide the human deoxyribonucleic acid (DNA) code for an increasing number of specific diseases, and the application of this data to human disease is still ongoing.1,2 Every aspect of disease management may eventually be affected by decoding its genetic basis. Prenatal diagnosis and carrier state detection should allow early diagnosis so that treatment can be directed at prevention with targeted protein treatments or gene therapy prior to the manifestation of the disease.3,4 A clear understanding of genetic principles begins to allow the applications of molecular genetics to identify genes associated with disease, to clone or isolate those genes, to identify the gene product and the key pathologic events, and finally to design treatment strategies.5
The human genome consists of 22 paired chromosomes and one pair of sex chromosomes, X and Y. Females have two copies of the X chromosome, whereas males exhibit an XY genotype. The Y chromosome carries information that determines secondary male characteristics. Since males have only one X chromosome, recessive genes on the X chromosome in males are expressed phenotypically. Females with one normal allele and one disease allele are called carriers.6 The Lyon hypothesis predicts that one of the X chromosomes in a female is randomly inactivated in somatic cells early in embryogenesis.7 This inactivation may allow the display of some signs of the disease in carrier females. Diseases carried on the X chromosome are, therefore, genetically linked to the sex of the patient.
The X chromosome also has a disproportionately large number of genes coding for ocular traits8 (Table 1, Fig. 1). Given the linkage to the sex of the patient, pedigree analysis may identify some diseases carried on the X chromosome. In X-linked recessive diseases, pedigree analysis will reveal that more males are affected than females, and affected fathers pass the carrier state to 100% of their female offspring and none of their male offspring. Furthermore, both male and female offspring of a carrier female have a 50% possibility of inheriting the affected allele. Carrier females are unaffected, but may show variable signs of the disease9,10,11 (Fig. 2). X-linked dominant disease is rare, but identifiable by pedigree analysis. Females are affected. There are approximately twice as many affected females as males because females carry a larger percentage of all X chromosomes. The male genotype is often lethal, but if it is viable, affected males transmit the trait to all of their female offspring and to none of their male offspring. Affected females have a 50% probability of passing the disease allele to both their male and female offspring9,10,11 (Fig. 3). These inheritance patterns may allow the localization of the disease gene to the X chromosome and are important for genetic counseling. Isolation of a specific gene may employ several of the tools of molecular genetics, including restriction endonucleases and gel electrophoresis.3
TABLE 1. X-linked Diseases With Ocular Manifestations | |
|---|---|
|
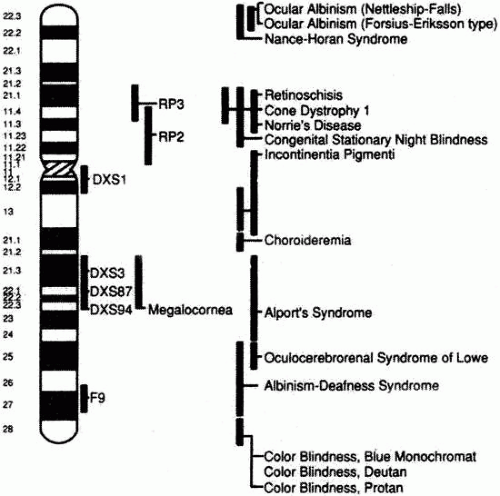 Fig. 1. Idiogram of the X chromosome showing the approximate locations of linkage markers and several X-linked ocular diseases. RP, retinitis pigmentosa; DXS, F9, known X-chromosome linkage markers. (Modified from Mackey DA, Buttery RG, Wise GM et al: Description of X-linked megalocornea with identification of gene locus. Arch Ophthalmol 109:829, 1991.) |
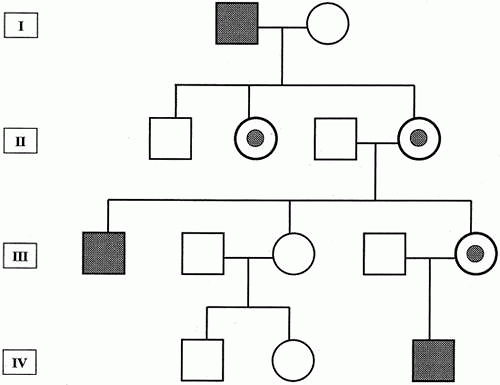 Fig. 2. Pedigree showing typical X-linked recessive inheritance. Generally, asymptomatic female carriers inherit traits from affected males. |
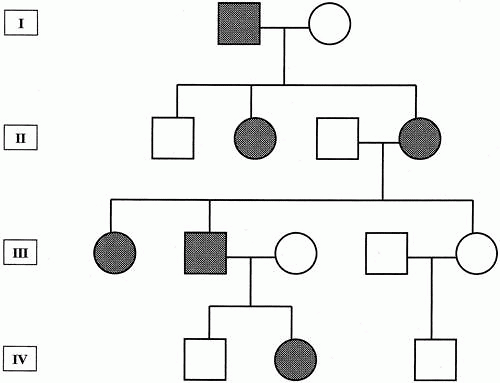 Fig. 3. Pedigree showing typical X-linked dominant inheritance. Affected males pass the trait to all their female offspring but to none of their male offspring. |
A specific restriction endonuclease enzyme will cleave DNA at a particular recognition site for that enzyme, cutting the DNA into manageable fragments in a predictable and reproducible manner.12 Because mutations in genes may be caused by base pair changes, deletions, or rearrangements of the genetic material, the genetic sequence in a mutation will be modified. If the modification occurs within a recognition site for a restriction endonuclease or includes an insertion or deletion of genetic material adjacent to a recognition site, the fragment lengths produced by the restriction endonuclease will differ from the nonmutated DNA. Restriction endonucleases, therefore, may be used to identify these mutations. The different-size fragments are referred to as restriction fragment length polymorphisms (RFLPs). Gel electrophoresis of the fragments allows the separation of biological molecules based on their size or secondary and tertiary structure. The technique can be tailored to exploit the characteristic of interest by manipulating the components of the gel.
Restriction endonucleases and gel electrophoresis are combined in Southern blot analysis. The total human genome is digested with a particular restriction endonuclease. The fragments are then separated by gel electrophoreses and transferred to a paper medium from the gel. A DNA probe, which is a unique sequence of DNA of interest labeled with either a radioactive or other type of marker, is hybridized with the paper to look for a specific sequence of DNA. Southern blot analysis has many applications, including the search for gene deletions and rearrangements in a pedigree.
These molecular genetic techniques are also employed in determining the location of a gene within the X chromosome. The location of a gene of interest can be estimated by using linkage analysis. Linkage analysis employs specific markers whose position on the chromosome is known relative to each other. These markers must exhibit many different alleles in the population to allow a specific allele to be traced through the pedigree. By comparing the inheritance of these markers to the inheritance of the disease of interest in a pedigree, linkage can be ascertained. Linkage implies that the disease gene is present near the same chromosomal locus as the marker. The closer together two genes are on a chromosome, the less likely they are to recombine during meiosis. Thus, if the disease gene and the marker are close together, they are not likely to separate and will be inherited together. This linkage can be detected with Southern blot analysis. For example, a RFLP is a type of linkage marker. If an endonuclease cleavage site is near a gene of interest, then RFLPs are inherited along with the disease. When screened, patients with the disease will also inherit the same RFLP pattern.13,14 Another type of linkage marker is a variable number of tandem repeats (VNTRs). VNTR polymorphisms are regions of DNA where a particular short sequence of DNA is repeated. The number of repeats is highly variable and can be detected in genetic analysis. A third possible marker is a dinucleotide repeat or “CA” repeat.13 If these sequences are located close to a gene of interest, then both the gene of interest and the number of repeats will be inherited together. This pattern can be detected in offspring. Testing for these linkage markers can be used in prenatal diagnosis and carrier state detection.
Linkage analysis suggests the approximate location of the disease gene on a chromosome. Identification of the specific gene may be by the candidate gene approach or the positional cloning of all the genes in the region until the disease gene is isolated. A candidate gene must either be expressed in the disease tissue or have a known or suspected function in the diseased tissue. In the positional cloning method, there are no assumptions made about the nature of the gene product or its function, but this approach is labor intensive and time consuming. On the other hand, identification of a new gene product may lead to insights into the physiology of diseases.
Upon identifying a gene, the functional coding regions of the gene, or exons, are separated from the noncoding introns. Mutations are alterations in the genetic code. Mutations can be classified in the following categories: missense mutations, nonsense mutations, frame shift mutations, and splice site mutations. Missense mutations change a single amino acid in the protein product due to substitution of a nucleotide in the coding sequence. Nonsense mutations result in a stop codon that leads to a truncated protein. Frame shift mutations are caused by nucleotide deletions or insertions that completely alter the protein product. Splice site mutations affect the transcription of DNA to RNA by altering the exon-intron boundaries of the gene. Base pair mutations may be designated either by the nucleic acid substitutions followed by the numeric position within the gene product, such as GGC-to-GAC base 164, or by the amino acid substitution at the numeric position in the gene product, such as C203R.
Focus on identifying a gene product may lead to a greater understanding of the pathogenesis of the disease in question. Antibody labeled to the gene product can then be used to isolate the location of the protein in the ocular tissue. The localization may help determine the function of the protein. In the future, gene product identification may also allow specific target protein treatment.
The X chromosome carries many genes for ocular diseases as well as for systemic diseases that have ocular manifestations. The systemic diseases are discussed elsewhere in this book. This chapter focuses on the more frequent and severe ophthalmic diseases, including choroideremia, color vision deficits, megalocornea, Norrie’s disease, ocular albinism, retinitis pigmentosa, X-linked cataracts, and X-linked juvenile retinoschisis. Discussion focuses on the influence of molecular genetics on diagnosis and treatment.
CHOROIDEREMIA
Choroideremia is a bilateral progressive dystrophy of the retinal pigment epithelium (RPE), photoreceptors and choroid that results in progressive loss of peripheral vision with sparing of central vision until late in the disease.15 Initially described by Mauthner in 1872,16 the disease was thought to be a result of a congenital absence of the choroid. However, it is now apparent that absence of the choroid is a late feature in the disease process and is not congenital. The prevalence varies with the population studied but has been estimated to be 1:100,000.10,17
Most affected males with choroideremia present with symptoms of nyctalopia in the first decade of life.18 Progressive peripheral visual loss ensues. By the third to fourth decade, there is severe constriction of the visual field with anular scotomas or tunnel vision on visual field testing.19,20 Central visual acuity is typically reduced to 20/200 or worse by age 50. Progressive myopia and disorders of color vision have also been reported. Choroideremia is generally isolated to the ocular structures, but sensorineuronal hearing loss has been reported in some affected individuals.17 Choroideremia displays wide variability in disease severity and rate of progression.
Clinically, funduscopic changes are initially apparent in the mid-peripheral fundus, with progression toward the macula and the far-periphery (Fig. 4A). These changes include RPE stippling and atrophy, with focal loss of choriocapillaris and prominence of the underlying choroidal vessels surrounding the optic disc (Fig. 4B). Early in the disease process, areas of skip lesions are clearly evident on fluorescein angiography. Additionally, scattered intraretinal pigment clumps at the equator and midperiphery may be noted. At this early stage, abnormal light and dark adaptation can be detected, and the electroretinogram (ERG) reveals reduced rod responses. Patches of chorioretinal degeneration occur near the equator and eventually advance, with loss of the normal choroidal pattern and diffuse RPE and choroidal atrophy except in the macular region. However, no intraretinal pigment migration occurs, as in retinitis pigmentosa.21 The macular area is the last to be affected and is usually the only area of normal tissue left in the late stages of the disease. In the final stages of choroideremia, the scotopic ERG is unrecordable.16,18
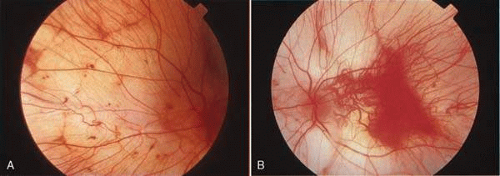 Fig. 4. A. Choroideremia in a male patient. There is loss of the retinal pigment epithelium and prominence of the choroidal pattern in the periphery. There is sparing of the macular region. B. Portions of the retinal pigment epithelium under the fovea still are intact in the late stages of choroideremia. (A and B courtesy of J. M. D. Gass, M.D.) |
Female carriers rarely may manifest a milder version of the disease but typically are asymptomatic, although they may exhibit some of the fundoscopic changes in the retina. The typical appearance involves a mottled mosaic fundus with linear retinal pigmentation and punctate areas of pigment epithelial atrophy.16
Histopathologically, loss of the choroid, RPE, and outer retinal layers are evident in eyes of patients with advanced choroideremia.22 In histological samples with intact retinal photoreceptors, the RPE and its basement membrane are duplicated and thickened. One copy is pigmented and the other nonpigmented.23 Female carriers have also shown diffuse abnormalities of the RPE on histopathologic examination. These abnormalities include patchy depigmentation of the RPE, coarse pigment granularity, and peripheral clumps of pigment. Scanning electron microscopy reveals pleomorphic RPE cells with loss of polygonal structure and villi. There is preservation of the underlying choriocapillaris and Bruch’s membrane.24 These observations support the hypothesis that the abnormal RPE cell is the primary feature in choroideremia, with secondary effects on the retina and the choroid.24,25,26 However, controversy exists as to the actual site of pathogenesis, because several other reports have hypothesized an abnormality in the choriocapillaris.27,28,29 In addition, rod photoreceptors have recently been identified as a possible primary site of degeneration in this disease.30
Choroideremia is inherited in an X-linked recessive pattern.31 The genetic focus for choroideremia, now known as the choroideremia (CHM) gene, has been identified by linkage analysis and cloning to Xq21.2.21,32,33,34 The CHM gene consists of 15 exons and encodes a ubiquitously expressed protein, the Rab escort protein-1 (REP-1) of geranylgeranyl (GG) transferase.35,36 This enzyme facilitates isoprenylation of cysteine residues in Rab proteins, a family of guanosine triphosphate (GTP)–binding proteins that regulate vesicular traffic. REP-1 is one of the two components of Rab GG transferase. REP-1 binds to Rab proteins and allows the second catalytic component of Rab GG transferase to transfer the GG moiety to the Rab protein. As a consequence of this chemical reaction, the Rab protein becomes more hydrophobic and integrates itself into the cell membrane where it is activated to a GTP-bound state. The Rab protein is then able to perform vesicular transport and membrane regulatory functions.24,37 A marked deficiency in the functional activity of Rab GG transferase was found in lymphoblasts of choroideremia patients. Residual enzyme activity has been detected in patients with choroideremia. This may be due to partial compensation by another gene—choroideremia-like (CHML)/REP-2—located on chromosome 1. It is hypothesized that CHML/REP-2 cannot fully compensate for deficiency of REP-1. The ocular structures, in particular, may be more sensitive to the presence of REP-1 for adequate activity of Rab GG transferase, explaining why the clinical features of choroideremia are confined to the eye.17 The exact site of pathogenesis has not yet been identified.
Mutations that alter the CHM gene and result in the truncation or absence of the CHM/REP-1 protein have been implicated in choroideremia. These mutations include nonsense, frameshift, and splice-site mutations. Missense mutations have generally not been identified, with one possible exception.38 Most mutations eventually lead to the introduction of a premature stop codon which results in a truncated or absent gene product.39,40,41 No specific exon in the CHM gene is more susceptible than others to mutations.42 Interestingly, the mutations described in European, Canadian and American families vary significantly from those reported in Japanese families. This implies an independent origin for these separate mutations.42
Prenatal diagnosis of choroideremia is performed during the 12th week of prenancy with indirect linkage analysis using a linked polymorphic DNA marker to analyze chorionic villi.43,44 The protein truncation test is another useful diagnostictool. It can be used to detect the alteration causing chorioderemeia n in unrelated patients by detecting truncated protein products.45 Choroideremia can also be confirmed by immunoblot analysis utilizing anti-REP-1 antibody. The absence of the REP-1 protein in peripheral blood samples is diagnostic of choroideremia.46
Despite better understanding of the molecular components of choroideremia, the exact pathogenesis of the disease is unknown and no treatment currently exists. Duncan and coworkers evaluated the effect of lutein supplementation in patients with choroideremia over a 6-month period. Supplementation led to an increase in serum lutein and macular pigment levels; however, no change in central vision was noted.48
COLOR VISION DEFECTS
Three classes of cone photoreceptors encode normal color vision. Red, green, and blue cone opsin pigments are sensitive to long (562 nm), middle (530 nm) and short (420 nm) wavelengths, respectively.48 The absorbance spectrum of these pigments is maximum at the wavelength specified above but is broad enough to cover the entire spectrum of visible light. Each photoreceptor cone contains only one pigment opsin in its outer segment. A color is detected by the mixture of signals from these pigment opsins, allowing the perception of the normal array of colors.
Since the system of color detection is a three-pigment system, normal color vision is trichromatic. If one of the pigments is absent, the person is known as a dichromat. A further descriptor is added to denote the specific opsin absent. For example, a protanopic dichromat lacks the red pigment, whereas a deuteranopic dichromat lacks the green pigment. A tritanopic dichromat, lacking the blue pigment, is rare. Approximately, 8% of Caucasian males and 0.5% of Caucasian females exhibit some abnormality of red/green color detection.49 Color deficits are equal in each eye and nonprogressive. The majority of persons with red/green deficiencies have abnormally functioning pigment rather than complete absence of pigment. The abnormal pigment has an altered absorption spectrum. Deuteranomalous trichromats have abnormal green pigment and are five times more common than protanomalous trichromats with abnormal red pigment. If both red and green pigments are missing, the individual has blue-cone monochromacy and essentially lives in a monochrome world.
There are several different methods for testing color vision. These tests reveal the axis of color deficiency, and the severity of the abnormality. The most common test is the Ishihara pseudoisochromatic plates assay. The 38-plate edition of the Ishihara diagnostic plates correctly classified 98.7% of patients.50 The Rayleigh color match test with an anomaloscope is commonly used for research purposes and can detect not only the axis of color deficiency but also abnormalities in maximum absorption spectrum. The anomaloscope projects a yellow light onto half of a screen. Patients are asked to adjust the ratio of a mixture of red and green light projected on the other half of the screen until both halves of the screen match. Persons with normal color vision use a highly reproducible ratio of red to green light. Dichromats match the yellow standard with any ratio of red and green light, including red or green light alone. Anomalous trichromats require a higher proportion of either red or green light to make the match. Other tests include the Farnsworth D-15 and the Farnsworth-Munsell 100-hue tests. In all test methods, sufficient illumination must be maintained for proper testing.
The understanding of color vision deficits was revolutionized by the cloning of the three visual pigments.51 These pigments are transmembrane proteins that confer color sensitivity to the cone photoreceptors. Color pigments are composed of 348 to 364 amino acids.51 Each pigment has seven hydrophobic sections arranged in seven helices that span the photoreceptor membrane.52 These transmembrane segments are linked by extracellular hydrophilic loops. The helices create a pocket for the binding of retinal. Retinal becomes activated by a photon of light and changes from 11-cis retinal to the all-trans retinal, causing activation of the photoreceptor. The maximum activation depends upon the wavelength sensitivity determined by the particular pigment protein to which the retinal is bound. The red and green opsin pigments differ at only 15 amino acid sites.52 These differences are located within the membrane helices and in close proximity to the retinal binding site. The electronic milieu surrounding the retinal is important in determining the ease of transformation of the retinal.53 This milieu is determined by the charges of the amino acids of the visual pigment. Amino acid substitutions that alter the net charge may promote ease of transformation, which changes the maximum wavelength of activation of the photoreceptor.
The three classes of pigments vary in length, but the helices and the extracellular loop structures are highly conserved. Two cysteine residues within the first and second extracellular loops form a disulfide bridge essential for the proper folding of the protein. These cysteines are highly conserved.52 Alterations in critical amino acid sequences can cause mutations that render a pigment inactive or change the maximum absorbance spectrum of the pigment.
The blue pigment gene is located on chromosome 7, and the red and green pigment genes are located on the X chromosome, Xq22-q28. The red and green pigment genes are arranged in a head-to-tail tandem array. Each gene consists of six exons separated by long noncoding introns. The red gene is approximately 15.1 kilobases (kb) in length and the green gene is 13.2 kb. The primary codons involved in the spectral tuning of these two pigments are located in exon 3 (site 180) and exon 5 (sites 277 and 285).51,54 A single, red pigment gene is located at the 5′ position of the pigment array, followed by one or more green pigment genes.55
A locus control region is located upstream of the red pigment gene. This region is essential for the expression of the particular pigment in the cone outer segment. Individuals with multiple green pigments have only one green pigment expressed.56 In one study, photoreceptors of 92% of individuals expressed the first two pigments arranged in the color gene array.57 Although it is generally accepted that the green gene expressed is usually the most proximal one in the gene array,58 other studies present data that conflict with this hypothesis.59
The red and green pigment genes are susceptible to mispairing during meiosis owing to the similarity in sequencing and their close proximity on the X chromosome. This leads to unequal crossing over, with deletion of a gene from one chromosome and its addition to the other. If the crossover occurs within one of the pigment genes, a hybrid gene is formed. These hybrid genes are responsible for anomalous color vision.52 Since sites 277 and 285 are important in the determination of wavelength maximum absorbance and are in close proximity, they are likely to remain together in hybrids. Exon 5, therefore, largely determines the maximum absorption spectrum of the visual pigment.60 Hybrids with the red codons in exon 5 but the green codons in other exons can have a maximum absorption shifted from the 562 nm expected of red pigment opsins to between 545 and 557 nm.61 Protan defects have been associated with red-green hybrid genes, and deutan defects with either green pigment gene deletion or a green-red hybrid gene.62,63,64
Several point mutations in the pigment genes have been well described. Mutations substituting critical amino acids result in pigments with no absorbance of light or an altered absorbance spectrum.65,66 One mutation, C203R, changes the cysteine required for the disulfide bridge. This destabilizes protein folding and renders the mutated protein unable to function.65 If both the red and green pigment genes are nonfunctional, blue cone monochromacy results.67 Because of the X-linked recessive nature of color blindness, females do not exhibit the color deficiency phenotype, but carrier females can be detected by genetic analysis.68 These female carriers do show some reduced color purity discrimination when tested with more extensive methods69 and may express more than three pigments owing to random inactivation of one X chromosome.70
Despite the better understanding of the specifics of the visual pigment genes, it remains difficult to predict a person’s phenotype for color vision. Normal individuals often possess green-red hybrid genes,62 and individuals with color deficits can possess normal pigment genes. Lack of correlation of the genotype with the phenotype found on testing may be due to selective gene expression in the red/green array.58,62,71 The locus control region determines which gene is expressed in each photoreceptor. The exact gene expressed may also depend on the degree of folding or looping of the X chromosome that brings the locus control region into contact with a single promoter region.62 ERG has been proposed to detect the spectral sensitivity of the photoreceptors to more accurately determine which genes are expressed.72 Nevertheless, other factors, such as limitations in testing methods, may influence color detection. Local ocular factors may also influence maximum spectral absorbance, such as preretinal absorbance of light, variation in photopigment optical density, and other optical effects within the photoreceptor.64 At present, although it is not possible to predict an individual’s maximum absorbance spectrum for each pigment based on the genotype,56 in general, the principles governing color detection are understood.
MEGALOCORNEA
Megalocornea is a rare congenital condition characterized by bilateral symmetric corneal enlargement, with the horizontal diameter greater than 13 mm by 2 years of age. Although there are both autosomal dominant and autosomal recessive variants of megalocornea, the most common hereditary pattern is X-linked recessive, which was first described in 1914 in a kindred of 17 affected males.73 The differential diagnosis of megalocornea includes congenital glaucoma, ectopia lentis et pupillae,74 congenital miosis,75 and Reiger’s anomaly.76
The clinical characteristics of X-linked megalocornea include a cornea with normal central topography and with corneal enlargement occurring at the limbus.77 The corneal horizontal diameter ranges between 13 and 16.5 mm (Fig. 5). The corneal endothelial cell count, density, and morphologic features are normal. The cornea has normal thickness and translucency. Importantly, megalocornea occurs in absence of elevated intraocular pressure. The visual acuity is usually preserved. The other clinical characteristics include enlarged anterior chamber depth, prominent iris processes, increased pigmentation in the anterior chamber angle, Kruckenberg’s spindle, iris stromal hypoplasia, iridoensis, miosis,78 posterior subcapsular cataracts, and minimal myopia with less than 2 diopters (D) of with-the-rule astigmatism.79,80
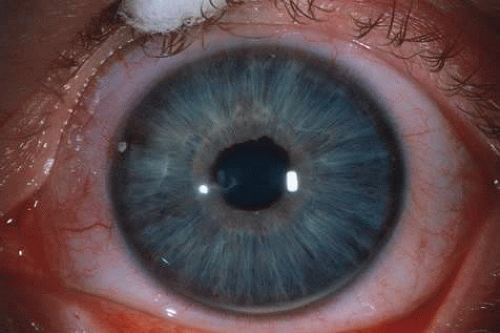 Fig. 5. Megalocornea. (Courtesy of Dr. E Cohen.) |
Megalocornea is generally nonprogressive, although in one pedigree the onset of arcus lipoides and mosaic corneal dystrophy occurred in early adulthood.81 The arcus lipoides occurs in the absence of hypercholesterolemia. Female carriers of megalocornea may exhibit a slight increase in corneal diameter,82 although the majority of carriers show no signs of megalocornea.81
Megalocornea is generally an ocular isolated finding. Associations with other genetic syndromes do not occur with this X-linked disease. Various other disorders that exhibit megalocornea include Marfan syndrome,83 Sotos syndrome (cerebral gigantism),84 X-linked ocular albinism,85 ichthyosis,86 nonketotic hyperglycinemia,87 and Rieger-type syndrome.88
Megalocornea is typically diagnosed by its clinical features and pedigree analysis. However, in infants examination under anesthesia maybe required to differentiate megalocornea from congenital glaucoma. Biometric data may be obtained to confirm the large corneas, identify the short radius of the cornea, and measure the anterior chamber and vitreous depth. The anterior chamber depth in megalocornea is greater than the mean plus two standard deviations, and the vitreous depth is short.89 The short corneal radii give a globular shape to the cornea, which is pathognomonic for megalocornea if the cornea is less than 15 mm in diameter. Intraocular pressure is normal and specular microscopy reveals normal endothelial cell density, in contrast to the diminished densities in congenital glaucoma.
The gene locus for X-linked megalocornea (XLM) has been linked to the X chromosome markers DXS87 and DXS94.90 Linkage analysis has isolated the gene locus to the region of Xq21-Xq22.80 XLM has not yet been isolated, nor has its gene product been identified.
The etiology of megalocornea has been proposed to be an abnormal growth rate of the ectoderm of the optic cup embryologically, resulting in enlargement of the ciliary ring.91 This enlargement leads to enlargement of the entire anterior segment of the eye and shortening of the posterior segment, which is seen in megalocornea. The gene product of XLM is, therefore, hypothesized to control the growth of the ectoderm into the optic cup. Further studies are needed to isolate the gene and gene product. Since the disease is proposed to occur in early embryogenesis, carrier state detection and genetic counseling would be the aim of molecular diagnosis.
NORRIE’S DISEASE
Norrie’s disease (ND) is a bilateral retinal dysplasia with degenerative and proliferative changes in the retina and vitreous that result in retinal detachment and progressive atrophy of the eyes. It was first described in 1927 in affected males who subsequently developed complete retinal detachments.92 The following ocular manifestations have also been reported: falciform-fold formation in the retina, retinal pigment epithelium proliferation, persistent hyperplastic primary vitreous, vitreous hemorrhage, iris synechiae, glaucoma, and within the first few months of life, the formation of cataracts and corneal opacities.93,94,95 The ophthalmic manifestations vary, and intrafamilial phenotype variability is high.96
The systemic manifestations of ND include mild to severe mental retardation in one third of cases.93 Another third exhibit a progressive decline in cognitive function, often with psychotic features. The majority of affected infants have normal cognitive development until 2 years of age, and then gradually begin losing skills through adulthood. The remaining third of children are cognitively normal. Warburg reported sensorineuronal hearing loss in one third of all children that began in the third decade of life.93 Although ND has also been reported to have characteristic facies including fine features, narrow nasal bridge, hypotelorism, flattened malar region, thin upper lip, large ears,97 and other features such as microcephaly, cryptorchidism and limb anomalies,98,99 other series of patients with ND have not exhibited these systemic features.100 The differential diagnosis of ND includes all causes of leukocoria in infancy including retinoblastoma, persistent primary hyperplastic vitreous, familial exudative vitreoretinopathy, and Coat’s disease. The diagnosis of ND can be confirmed with genetic testing for the ND gene.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


