Chapter 75 Vogt–Koyanagi–Harada Disease
Introduction and historical aspects
Vogt–Koyanagi–Harada (VKH) disease is a bilateral granulomatous uveitis often associated with exudative retinal detachment and with extraocular manifestations, such as pleocytosis in the cerebrospinal fluid and, in some cases, vitiligo, poliosis, alopecia, and dysacusis.1
Poliosis associated with ocular inflammation was first described by Ali-ibn-Isa, an Arab physician who lived in the 1st century AD (cited by Pattison).2 This association was reported by Schenkl in 1873,3 by Hutchinson in 1892,4 and by Vogt in l906.5 Harada described a primary posterior uveitis with exudative retinal detachments in association with cerebrospinal fluid pleocytosis.6
Three years later, in 1929, Koyanagi described six patients with bilateral chronic iridocyclitis, patchy depigmentation of the skin, patchy hair loss, and whitening of the hair, especially the eyelashes.7 This constellation of findings was termed “uveitis with poliosis, vitiligo, alopecia, and dysacusis.”7 Babel in 19328 and Bruno and McPherson in 1945 combined the findings of Vogt, Koyanagi, and Harada and suggested that these processes represent a continuum of the same disease,9 thereafter recognized as Vogt–Koyanagi–Harada syndrome.
When a patient presents with the ocular and the extraocular manifestations, the diagnosis of VKH is made with certainty. However, extraocular manifestations such as dysacusis and cutaneous changes are relatively rare, and the dermatologic changes mainly occur late in the course of the disease.1,10 Because of the variation in clinical presentations of VKH, the American Uveitis Society (AUS) in 1978 recommended the following diagnostic criteria: (1) the absence of any history of ocular trauma or surgery; and (2) the presence of at least three of the following four signs: (a) bilateral chronic iridocyclitis; (b) posterior uveitis, including exudative retinal detachment, forme fruste of exudative retinal detachment, disc hyperemia or edema and “sunset glow” fundus; (c) neurologic signs of tinnitus, neck stiffness, cranial nerve, or central nervous system disorders, or cerebrospinal fluid pleocytosis; and (d) cutaneous findings of alopecia, poliosis, or vitiligo.11
Since VKH manifestations vary depending upon the clinical course, a given patient may not initially present with the features required for the diagnosis of VKH by AUS criteria. Read and Rao recently evaluated the utility of the existing AUS criteria in 71 consecutive patients with VKH who were diagnosed based on the clinical features and the course of the disease, combined with fluorescein angiography with or without utrasonography in selected cases.12 The authors concluded that AUS criteria for diagnosis of VKH may not be adequate. Taking into account the multisystem nature of VKH and allowing for the different ocular findings present in the early and late stages of the disease, the First International Workshop on VKH proposed revised diagnostic criteria to include clinical manifestations at various stages of disease.13 These revised diagnostic criteria are summarized in Box 75.1.
Box 75.1
The revised diagnostic criteria proposed by the First International Workshop on Vogt–Koyanagi–Harada (VKH) disease*
B. Incomplete VKH disease (point 1 and either 2 or 3 must be present)
(Modified from Read RW, Holland GN, Rao NA et al. Revised diagnostic criteria for Vogt–Koyanagi–Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001;131:647–52.13)
In the past, constellation of these ocular signs and symptoms warranted the term “syndrome,” but in recent years the entity has been well characterized; thereafter the International Workshop on VKH adopted the term Vogt–Koyanagi–Harada disease.13
Epidemiology
The incidence of VKH is variable. It appears to be more common in Japan, where it accounts for 6.7% of all uveitis referrals.14 In the USA it accounts for 1–4% of all uveitis clinic referrals.
VKH tends to affect more pigmented races, such as Asians, Hispanics, American Indians, and Asian Indians.1,l5 In the USA there appears to be variability in the racial distribution of patients with VKH disease.1,11,16,17 In northern California VKH was seen mainly in Asians (4l%), followed by whites (29%), Hispanics (16%), and blacks (14%).17 In contrast, reports from southern California show that 78% of VKH patients were Hispanic while 3% were white, 10% were Asian, and 6% were black.1 A series reported from the National Institutes of Health (NIH) showed that 50% of VKH patients were white, 35% were black, and 13% were Hispanic.17 However, most of those patients reported in the NIH series had remote American Indian ancestry. Most studies report that women tend to be affected more frequently than men; however, Japanese investigators have not found such a female predilection.15 Most patients are in their second to fifth decades of life, but children may also be affected.1,18
Clinical description
Typical clinical features of VKH include bilateral panuveitis associated with exudative retinal detachment, meningism associated with headache and pleocytosis of cerebrospinal fluid, tinnitus or hearing loss, and cutaneous changes, such as alopecia, poliosis, and vitiligo. However, all of these extraocular features are rarely seen during the initial presentation, and the clinical features vary depending upon the stage of the disease as well as the effect of medical treatment. Presence of ocular and two or more extraocular features is considered as a complete form of VKH disease.13 Incomplete VKH disease includes bilateral typical ocular involvement plus either neurologic/auditory or cutaneous changes, whereas probable VKH disease is composed of just ocular manifestations.13 However, some of these probable VKH patients can develop cutaneous manifestations during the chronic or chronic recurrent stage of the disease.
The acute uveitic stage
This stage follows the prodromal phase and presents with blurring of vision in both eyes. One eye may be affected first, followed a few days later by the second eye. Despite a delay in symptoms, careful examination will reveal bilateral posterior uveitis. This uveitis consists of thickening of the posterior choroid with elevation of the peripapillary retinochoroidal layer, multiple serous retinal detachments (Fig. 75.1), hyperemia and edema of the optic nerve head.
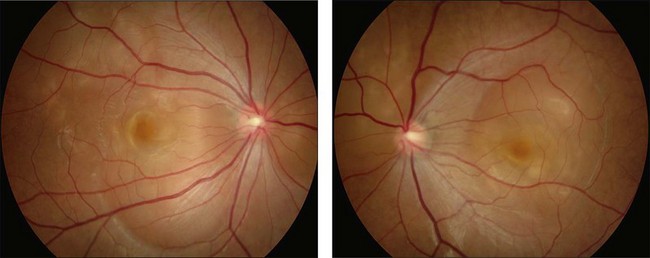
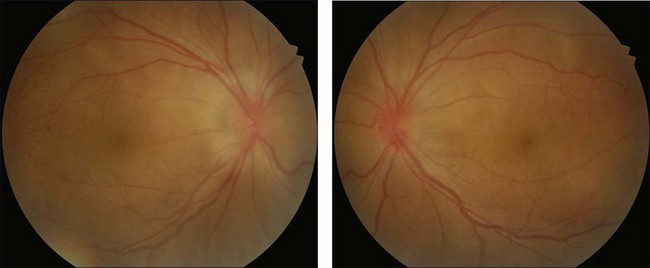
Rarely VKH disease can present with optic disc hyperemia and edema without serous retinal detachments (Fig. 75.2).
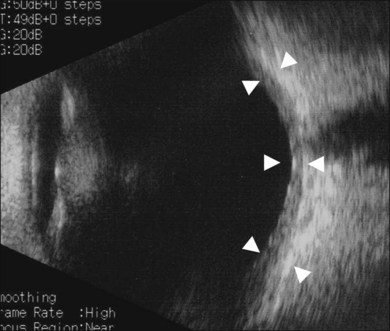
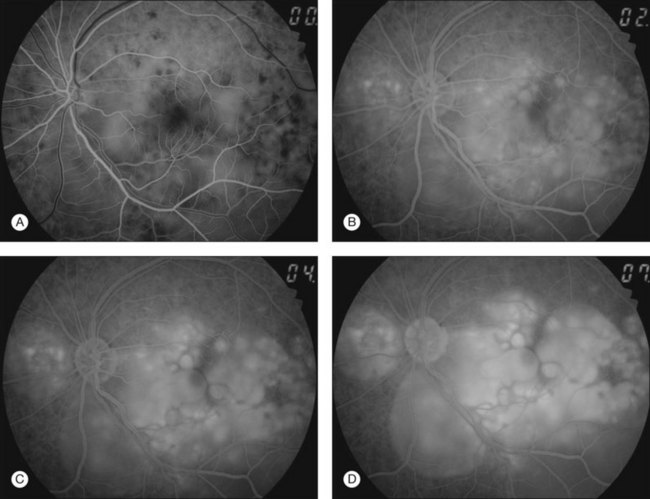
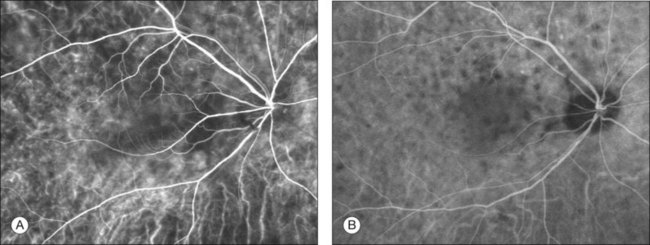
Thickened choroid can be detected by ultrasonography (Fig. 75.3). Alteration in the retinal pigment epithelium (RPE) associated with multifocal choroidal inflammation is easily observed with fluorescein angiography, which reveals hypofluorescent dots at the early phase followed by multiple focal areas of leakage and subretinal fluid accumulation at the late phase (Fig. 75.4).
Indocyanine green angiography (ICGA) (Fig. 75.5) could be useful to evaluate choroidal inflammatory changes such as early choroidal stromal vessel hyperfluorescence and leakage, and hypofluorescent dark dots at the level of the choroid.19,20
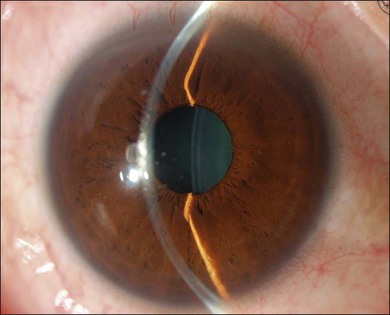
The inflammation eventually becomes diffuse, extending into the anterior segment and revealing the presence of flare and cells in the anterior chamber. Less commonly, mutton-fat keratic precipitates, small nodules on the iris surface and pupillary margin, may be observed;1 however, these anterior inflammatory changes are more common in the recurrent phase. The inflammatory infiltrate in the ciliary body and choroid may cause forward displacement of the lens iris diaphragm (Fig. 75.6), leading to acute angle closure glaucoma or annular choroidal detachment.21,22 These intraocular changes are typically bilateral; rarely, however, the process can be restricted to one eye.23
The chronic uveitic stage
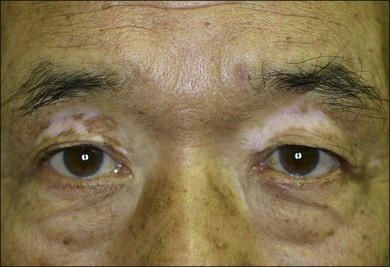
The chronic or convalescent stage occurs several weeks after the acute uveitic stage and is characterized by development of vitiligo (Fig. 75.7), poliosis, and depigmentation of the choroids. Perilimbal vitiligo, also known as Sugiura’s sign (Fig. 75.8), may develop at this stage among the patients who have melanosis at the palisade of Vogt, such as Japanese patients.1,13
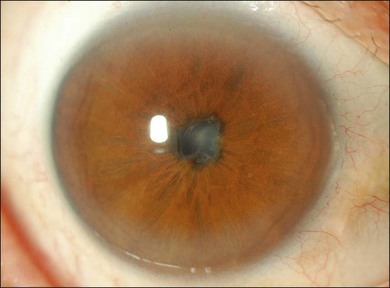
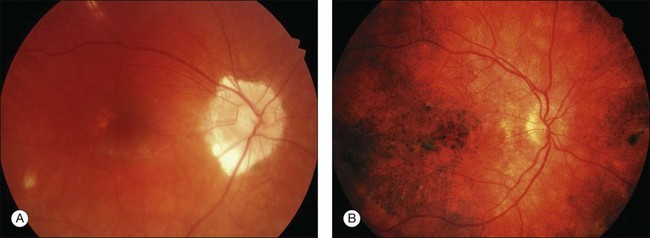
Choroidal depigmentation occurs a few months after the uveitic phase. This leads to the characteristic pale disc with a bright red–orange choroid known as sunset glow fundus (Fig. 75.9). In Hispanics, the sunset glow fundus may show foci of RPE changes in the form of hyperpigmentation or hypopigmentation. The juxtapapillary area may show marked depigmentation. At this stage small, yellow, well-circumscribed areas of chorioretinal atrophy may appear, mainly in the inferior midperiphery of the fundus. This convalescent phase may last for several months.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


