Vitreous Pathobiology and Anomalous Posterior Vitreous Detachment
J. Sebag
DEVELOPMENTAL AND INHERITED DISORDERS
Hyaloid Vascular System
The embryonic vascular system of the vitreous (vasa hyaloidea propria) attains its maximum prominence during the ninth week of gestation or 40-mm stage.1 Atrophy of these vessels begins posteriorly with dropout of the vasa hyaloidea propria, followed by the tunica vasculosa lentis, the embryonic vessels of the lens. Recent studies2 detected the onset of apoptosis in the endothelial cells of the tunica vasculosa lentis as early as day 17.5 in the mouse embryo. At the 240-mm stage (seventh month) in the human, blood flow in the hyaloid artery ceases.3 Regression of the vessel itself begins with glycogen and lipid deposition in the endothelial cells and pericytes of the hyaloid vessels.2 Endothelial cell processes then fill the lumen and macrophages form a plug that occludes the vessel. The cells in the vessel wall then undergo necrosis and are phagocytized by mononuclear phagocytes.4 Gloor5 claimed that macrophages are not involved in vessel regression within the embryonic vitreous but that autolytic vacuoles form in the cells of the vessel walls, perhaps in response to hyperoxia. Interestingly, the sequence of cell disappearance from the primary vitreous begins with endothelial and smooth muscle cells of the vessel walls, followed by adventitial fibroblasts and lastly phagocytes,6 consistent with a gradient of decreasing oxygen tension from within the blood vessel lumen out to the perivascular adventitia. Recent studies7 have suggested that vasa hyoidea propria and the tunica vasculosa lentis regress via apoptosis and that macrophages are important in this process. Subsequent studies by a different group8 confirmed the importance of macrophages in promoting regression of the fetal vitreous vasculature and further characterized these macrophages as hyalocytes.
Understanding what stimulates regression of the hyaloid vascular system would have great potential in enabling new approaches to the treatment (and perhaps even prevention) of neovascular disorders of the eye, such as proliferative diabetic retinopathy and exudative age-related macular degeneration. Indeed, inhibiting angiogenesis could also significantly improve the management of nonophthalmic disorders such as metastatic carcinoma. Studies have identified a protein native to the vitreous that inhibits angiogenesis in various experimental models.9,10 Activation of this protein and its effect on the primary vitreous may be responsible for the regression of the embryonic hyaloid vascular system as well as the inhibition of pathologic neovascularization in the adult. Mitchell et al.2 point out that the first event in regression of the hyaloid vasculature is endothelial cell apoptosis and propose that lens development separates the fetal vasculature from vascular endothelial growth factor-producing cells, decreasing the levels of this “survival” factor for vascular endothelium, inducing apoptosis. Following endothelial cell apoptosis there is loss of capillary integrity, leakage of erythrocytes into the vitreous, and phagocytosis of apoptotic endothelium by hyalocytes, the resident macrophages, or tissue histiocytes, of vitreous. Meeson et al.11 proposed that there are actually two forms of apoptosis that are important in regression of the fetal vitreous vasculature. The first (“initiating apoptosis”) results from macrophage induction of apoptosis in single endothelial cell(s) of an otherwise healthy capillary segment with normal blood flow. The isolated dying endothelial cell(s) project into the capillary lumen and interfere with blood flow. This stimulates synchronous apoptosis of downstream endothelial cells (“secondary apoptosis”) and ultimately obliteration of the vasculature. Removal of the apoptotic vessels is achieved by hyalocytes.
Pathology
Regression of the hyaloid artery and usually occurs completely and without complications. Persistence of the hyaloid vascular system occurs in 3% of full-term infants but in 95% of premature infants12 and can be associated with prepapillary hemorrhage.13 Anomalies involving incomplete regression of the embryonic hyaloid vascular system occur in more than 90% of infants born earlier than 36 weeks of gestation and in over 95% of infants weighing less than 5 lb at birth.14 There is a spectrum of disorders resulting from persistence of the fetal vasculature15.
Mittendorf Dot
Mittendorf dot is a remnant of the anterior fetal vascular system located at the former site of anastomosis of the hyaloid artery and tunica vasculosa lentis. It is usually inferonasal to the posterior pole of the lens and is not associated with any known dysfunction.
Bergmeister Papilla
Bergmeister papilla is the occluded remnant of the posterior portion of the hyaloid artery with associated glial tissue. It appears as a gray, linear structure anterior to the optic disc and adjacent retina and does not cause any known functional disorders. Exaggerated forms can present as prepapillary veils.
Vitreous Cysts
Vitreous cysts are generally benign lesions that are found in eyes with abnormal regression of the anterior16 or posterior17 hyaloid vascular system; otherwise normal eyes;18,19 and eyes with coexisting ocular disease such as retinitis pigmentosa,20 and uveitis.21 Some vitreous cysts contain remnants of the hyaloid vascular system22 supporting the concept that the cysts result from abnormal regression of these embryonic vessels.23 However, one histologic analysis of aspirated material from a vitreous cyst purportedly revealed cells from the retinal pigment epithelium.24 Vitreous cysts are generally not symptomatic and thus do not require surgical intervention. However, argon laser photocoagulation has been employed, and a recent report25 described the use of neodymium:yttrium-aluminum-garnet (Nd:YAG) laser therapy to rupture a free-floating posterior vitreous cyst.
Persistent Hyperplastic Primary Vitreous
Persistent hyperplastic primary vitreous (PHPV) was first described by Reese26 as a congenital malformation of the anterior portion of the primary vitreous appearing as a plaque of retrolental fibrovascular connective tissue. This tissue is adherent to the posterior lens capsule and extends laterally to attach to the ciliary processes, which are elongated and displaced centrally. Although 90% of cases are unilateral, many of the fellow eyes have Mittendorf dot or other anomaly of anterior vitreous development.27 A persistent hyaloid artery, often still perfused with blood, arises from the posterior aspect of the retrolental plaque in the affected eye. In severe forms there is microphthalmos with anterior displacement of the lens-iris diaphragm, shallowing of the anterior chamber, and secondary glaucoma. PHPV is believed to arise from abnormal regression and hyperplasia of the primary vitreous.27 Experimental data28 suggest that the abnormality begins at the 17-mm stage of embryonic development. The hyperplastic features result from generalized hyperplasia of retinal astrocytes and a separate component of glial hyperplasia arising from the optic nerve head.29 The fibrous component of the PHPV membrane is presumably synthesized by these astrocytes and glial cells.30 A case report31 with clinicopathologic correlation found that collagen fibrils in this fibrous tissue had diameters of 40 to 50 nm with a cross-striation periodicity of 65 nm. The investigators concluded that the collagen fibrils differed from those of the primary vitreous and suggested that they were either the result of abnormal metabolism by the cells that synthesize vitreous collagen, or arose from a different population of cells.
The retina is usually not involved in anterior PHPV. Indeed, previous studies32 have suggested that the anterior form is due to a primary defect in lens development and that vitreous changes are all secondary. This postulate has never been substantiated. There are rare instances of posterior PHPV31,33 in which opaque connective tissue arises from Bergmeister papilla and persistent hyaloid vessels (Fig. 1). These can cause congenital falciform folds of the retina and, if severe, can cause tentlike retinal folds leading on rare occasions to tractional and/or rhegmatogenous retinal detachment. Font et al.34 have demonstrated the presence of adipose tissue, smooth muscle, and cartilage within the retrolental plaque and suggest that PHPV arises from metaplasia of mesenchymal elements in the primary vitreous.
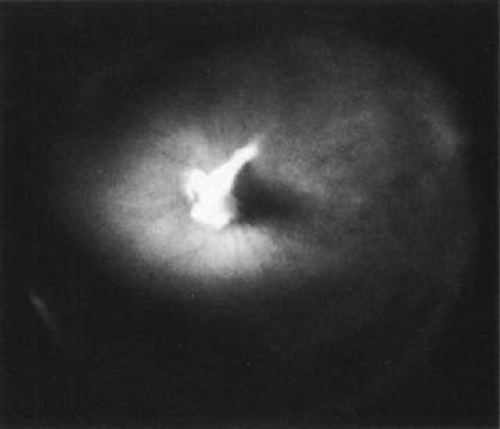 Fig. 1. Persistent hyperplastic primary vitreous (PHPV). A wide-angle fundus photograph of the left eye in a 10-year-old boy with the posterior form of PHPV demonstrates a fibrous stalk arising from the optic disc and extending into the central vitreous. (From Sebag J: The Vitreous: Structure, Function and Pathobiology. New York: Springer-Verlag, 1989:99, with permission.) |
Dominant Exudative Vitreoretinopathy
Dominant exudative vitreoretinopathy was first described in 1969 by Criswick and Schepens35 as a bilateral, slowly progressive abnormality of the vitreous and retina that resembles retinopathy of prematurity but with no history of prematurity or postnatal oxygen administration. Gow and Oliver36 identified this disorder as an autosomal dominant condition with complete penetrance. They characterized the course of this disease in stages ranging from posterior vitreous detachment with snowflake opacities (stage I), to thickened vitreous membranes and elevated fibrovascular scars (stage II), and vitreous fibrosis with subretinal and intraretinal exudates, ultimately developing retinal detachment due to fibrovascular proliferation arising from neovascularization in the temporal periphery (stage III). Plager et al.37 recently reported the same findings in four generations of three families, but found X-linked inheritance. Van Nouhuys38,39 studied 101 affected members in 16 Dutch pedigrees and five patients with sporadic manifestations. He found that the incidence of retinal detachment was 21%, all but one case occurring prior to the age of 30. These were all tractional or combined traction/rhegmatogenous detachments, and there were no cases of exudative retinal detachment. Van Nouhuys41 concluded that the etiology of dominant exudative vitreoretinopathy lies in premature arrest of development in the retinal vasculature, since the earliest findings in these patients were nonperfusion of the peripheral temporal retina with stretched retinal blood vessels and shunting with vascular leakage. Thus, Van Nouhuys considers dominant exudative vitreoretinopathy as a retinopathy with secondary vitreous involvement. However, Brockhurst et al.40 described that vitreous membrane formation begins just posterior to the ora serrata and that this precedes retinal vessel abnormalities, suggesting a vitreous origin to this disorder. Others41 suggest that there may be a combined etiology involving anomalies of the hyaloid vascular system and primary vitreous as well as retinovascular dysgenesis.
Retinopathy of Prematurity
Retinopathy of prematurity (ROP), a condition of premature infants, was first described in 1942 by Terry42 as retrolental fibroplasia. That term is no longer appropriate since it is actually a descriptive term of the pathology in advanced (stage V) cases of cicatricial ROP. We now identify acute stages of the disease that are not “retrolental” and do not have much “fibroplasia”.43 The pathogenesis of ROP44 begins with birth prior to complete maturation and development of the peripheral retina, followed by postnatal oxygen administration triggering retinal vasoconstriction with endothelial cell necrosis and vaso-obliteration in response to hyperoxia. After the discontinuation of supplemental oxygen, arterial pO2 levels return to normal and the obliterated (or at best highly constricted) vessels cause the peripheral retina they subserve to become ischemic and release neovascular growth factors. An alternative hypothesis of pathogenesis45 proposes that spindle cells in the immature peripheral retina are stimulated by excessive amounts of reactive oxygen species, whether related to oxygen therapy and subsequent relative hypoxia or other metabolic circumstances, to release angiogenic growth factors.
In either case the result is migration and proliferation of capillary endothelial cells that form new blood vessels at the posterior ridge of tissue between the vascularized and avascular retina. Neovascularization arises from the ridge that demarcates the developed posterior retina from the immature peripheral retina (Fig. 2). The new vessels grow into the vitreous body, similar to neovascularization in diabetic retinopathy,46 although they grow farther anteriorly and more exuberantly. This is perhaps due to the participation of cells of the ocular fetal vasculature whose apoptosis has been retarded or arrested by the presence of high levels of vascular endothelial growth factor.47
 Fig. 2. Gross and histologic (inset) appearance of an eye with ROP advancing from stage III to stage IV. Arrest of peripheral retinal development results in the lack of vitreous gel formation anteriorly (“Liquid”; “L” in the inset). Cell migration and proliferation along the interface between the gel vitreous posteriorly (“Gel”; “G” in the inset) and the liquid vitreous anteriorly produces the membranes in ROP that orient toward Weigert’s ligament anteriorly and induce traction detachment of the retina at the ridge demarcating posterior vascularized retina and peripheral avascular retina. (Courtesy of Maurice Landers, M.D.) |
There are no clearly identified vitreous changes during stages I and II of acute ROP, although this may simply be due to our present inability to detect48 such abnormalities. Indeed, the abundance of reactive oxygen species in the retina and vitreous of premature infants could induce widespread vitreous liquefaction.49 There are also likely to be localized areas of liquid vitreous, particularly at the periphery. At surgery for stage IV-A ROP with retinal detachment there is a “trough” in the periphery.50 This structure is most likely the consequence of underlying retinal immaturity in the periphery with consequent lack of typical gel vitreous synthesis, normally a Mueller cell function, overlying the immature retina. The liquid vitreous trough is probably present early in the natural history of disease but goes undetected by present vitreous imaging techniques.50 Such disruption of normal vitreous composition and structure probably alters a number of physiologic processes within the vitreous, including the ability of vitreous to inhibit cellular and vascular invasion.9,10,11 Furthermore the interface between posterior gel vitreous and peripheral liquid vitreous at the ridge causes vitreous traction to be exerted at the retinal ridge.
In stage III ROP, new blood vessels extend from the inner retina into the vitreous cortex. The cortex, overlying the rear guard of differentiated capillary endothelial cells, becomes opaque and contains linear, fibrous structures adjacent to a large pocket of liquid vitreous.51 In advancing from stage III to stage IV the neovascular tissue arising from the rear guard grows through the vitreous body toward Wiegert’s ligament on the posterior lens capsule.52 This configuration of neovascularization is probably the result of cell migration and proliferation along the anterior surface of the interface between gel vitreous posteriorly and liquid vitreous anteriorly (Fig. 2). Cells of the primary vitreous likely contribute to the formation of the dense central vitreous stalk and retrolental membrane seen in the cicatricial stage, since these cells could also undergo migratory and proliferative responses to intraocular angiogenic stimuli.
Vitreoretinal Dystrophy of Goldmann-Favre
Vitreoretinal dystrophy of Goldmann-Favre is an autosomal recessive condition first described in 1957 by Goldmann53 as consisting of vitreous abnormalities with peripheral retinoschisis and chorioretinal atrophy. Francois54 characterized the vitreous changes as syneresis (collapse), fibrillar degeneration with white strand formation, and punctate deposits. The retinal changes are pigmentary degeneration (with a different appearance from the bone spicules of retinitis pigmentosa), attenuated blood vessels, and microcystic degeneration of the macula and retinal periphery. Night blindness is an important feature, and the electroretinogram is markedly abnormal. Fishman and collaborators55 used fluorescein angiography to demonstrate cystoid macular edema (CME) and vascular occlusion with leakage in the peripheral retina corresponding to the areas of schisis. Schepens56 described that there is vitreous organization attached to the areas of schisis. Histopathologic studies by Peyman et al.57 showed attenuation of the outer nuclear layer and absence of photoreceptors. Based on the presence of normal retinal pigment epithelium and choriocapillaris, these investigators suggested that the etiology was a primary retinal degeneration. In this regard, Müller cell dysfunction could lead to the vitreous abnormalities as well as peripheral retinoschisis. It follows that in these cases, CME results from peripheral and central vitreous changes that occur in the absence of posterior vitreous detachment (PVD), thus inducing traction on the macula, as has been noted in cases of peripheral uveitis (or peripheral anterior vitreitis) with CME58,59 and in cases of aphakic macular edema.60,61 There can also be partial PVD with persistent attachment of vitreous fibrils to the macula (Fig. 3).
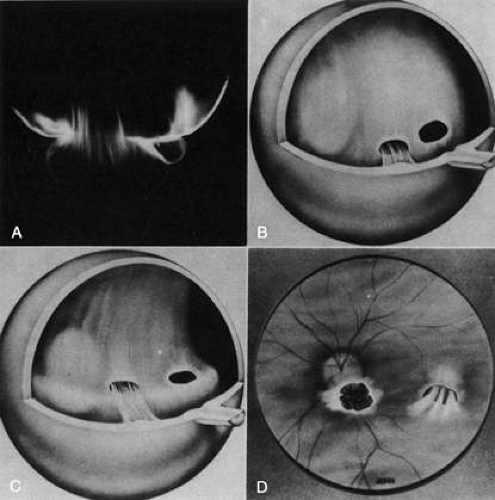 Fig. 3. Vitreomacular traction. A: Vitreous extrusion into the retrocortical (preretinal) space in a specimen from a middle-aged human. Vitreous can remain attached to the macula even in the presence of PVD. In such cases, vitreous can extrude through the premacular vitreous cortex, and fibers can insert into the macula. B-D: Artist’s rendition of this phenomenon. (Adapted from Jaffe NS: The Vitreous in Clinical Ophthalmology. St. Louis: CV Mosby, 1969; Jaffe NS: Vitreous traction at the posterior pole of the fundus due to alterations in the posterior vitreous. Trans Am Acad Ophthalmol Otolarynogol 71:642, 1967, with permission.) |
Vitreous Collagen Disorders
Collagen is one of the most important structural molecules in all connective tissues in the body, including vitreous. Thus, it is of interest to consider parallel phenomena occurring in the vitreous and connective tissues elsewhere, especially as related to collagen. For example, Gartner62 has pointed out the similarities between the intervertebral disk and vitreous, in which age-related changes with herniation of the nucleus pulposus were associated with presenile vitreous degeneration in 40% of cases. He proposed that a generalized connective tissue disorder resulted in disk herniation and presenile vitreous degeneration in these cases. Based on these findings, Gartner likened herniation of the nucleus pulposus in the disk to prolapse of vitreous into the retrohyaloid space by way of the posterior vitreous cortex following PVD (Fig. 3).
Maumenee63 has identified several different disorders with single gene autosomal dominant inheritance in which dysplastic connective tissue primarily involves joint cartilage. In these conditions there is associated vitreous liquefaction, collagen condensation, and vitreous syneresis (collapse). Since type II collagen is common to cartilage and vitreous, Maumenee suggested that the various arthro-ophthalmopathies may result from different mutations, perhaps of the same or neighboring genes, on the chromosome involved with type II collagen metabolism. In these disorders, probably including such conditions as Wagner disease,64 the fundamental problem in the posterior segment of the eye is that the vitreous is liquefied and unstable, tending to syneresis at an early age. However, there is no dehiscence at the vitreoretinal interface in concert with the changes inside the vitreous body, perhaps due to the fact that the internal limiting lamina of the retina is composed of type IV collagen. Thus, in these cases, abnormal type II collagen metabolism causes destabilization of the vitreous and results in traction on the retina that can lead to large, posterior tears and difficult retinal detachments.
Marfan syndrome is an autosomal dominant disorder featuring poor musculature, lax joints, aortic aneurysms, and arachnodactyly. Lens subluxation, thin sclera, peripheral fundus pigmentary changes, and vitreous liquefaction occur at an early age. The combination of myopia, vitreous syneresis, and abnormal vitreoretinal adhesions at the equator account for the frequency of rhegmatogenous retinal detachment due to equatorial or posterior horseshoe tears.58
Ehlers-Danlos syndrome has some similarities with Marfan syndrome, most notably joint laxity, aortic aneurysms, and an autosomal dominant pattern of inheritance, although there are as many as six types of Ehlers-Danlos patients, and the genetics of this condition are probably more heterogeneous than in Marfan syndrome. A further distinction from Marfan’s patients is the hyperelastic skin and poor wound healing of all connective tissues in Ehlers-Danlos patients, including cornea and sclera. Ocular manifestations include lens subluxation, angioid streaks, thin sclera, and high myopia due to posterior staphyloma. Vitreous liquefaction and syneresis occur at a young age. Vitreous traction causes vitreous hemorrhage, perhaps also due to blood vessel wall fragility, and retinal tears with rolled edges often cause bilateral retinal detachments.58
In 1965, Stickler et al.65 described a condition in five generations of a family that was found to be autosomal dominant with complete penetrance and variable expressivity. The features were a marfanoid skeletal habitus and orofacial and ocular abnormalities. Subsequent studies identified subgroups with short stature and a Weil-Marchesani habitus. The skeletal abnormalities now accepted as characteristic of Stickler syndrome are radiographic evidence of flat epiphyses, broad metaphyses, and especially spondyloepiphyseal dysplasia.66 Ocular abnormalities are high myopia, greater than -10 D in 72% of cases,67 and vitreoretinal changes characterized by vitreous liquefaction, fibrillar collagen condensation, and a perivascular latticelike degeneration in the peripheral retina believed to be the cause of a high incidence (greater than 50%) of retinal detachment.69 More recent studies68 have correlated specific gene defects with particular phenotypes, thereby enabling the classification of Stickler syndrome patients into four subgroups. Those patients with abnormalities in the genes coding for type II procollagen and type V/XI procollagen are the ones who have severe vitreous abnormalities.
Knobloch69 described a syndrome similar to Stickler syndrome with hypotonia, relative muscular hypoplasia, and mild to moderate spondyloepiphyseal dysplasia causing hyperextensible joints. The vitreoretinopathy is characterized by vitreous liquefaction, veils of vitreous collagen condensation, and perivascular latticelike changes in the peripheral retina. A recent case report70 of Knobloch syndrome describes a deficiency in endostatin as the cause for persistent fetal vasculature.
Myopic Vitreopathy
Considering how frequently myopia is associated with the aforementioned arthro-ophthalmopathies, it is of interest to consider whether myopia itself is due, at least in part, to vitreous collagen abnormalities. Since myopia is considered to be caused by both genetic and environmental factors,71 the genetic effects upon vitreous can be considered myopic vitreous dystrophy while the effects of environmental factors are appropriately termed myopic vitreous degeneration. The combined effects are best referred to as myopic vitreopathy. Since vitreous formation contributes to ocular growth by generating internal swelling pressure, it is likely that derangements in this process could alter the ultimate size of the eye. It is important to note that myopia is associated with enlargement not only in the longitudinal (optical) axis, but the vertical and horizontal axes, as well (Fig. 4).
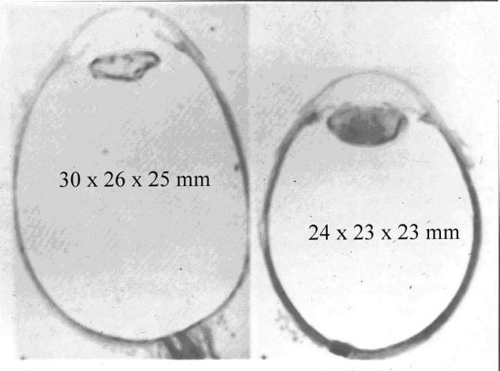 Fig. 4. Comparison of histologic cross sections of a myopic eye (left) with an emmetropic eye (right). Not only is the axial length greater in the myopic eye (30 vs. 24 mm), but so are the horizontal (26 vs. 23 mm) and vertical (25 vs. 23 mm) axes. This three-dimensional enlargement may be due in part to abnormal internal swelling pressure generated by deranged vitreous formation. (From Spencer WH: Ophthalmic Pathology. Philadelphia: WB Saunders, 1985, with permission.) |
Almost all of the growth of the eye after 2 or 3 years of age is due to expansion of the eye between the region of the ora serrata and the equator of the globe.72 Much, if not all, of the force inducing this expansion derives from the burgeoning growth of the vitreous body. In 1963, Coulombre et al.73 proposed that during development, vitreous growth was the major factor influencing intraocular pressure and eye growth. Subsequent studies74 showed that vitrectomized eyes with normal intraocular pressure had less ocular growth than controls. More recent studies75 showed that peripheral retinal ablation by cryopexy or laser photocoagulation in rabbits between 2 and 8 weeks of age caused significant reductions in axial length, equatorial diameter, corneal diameter, and total ocular volume. The investigators hypothesized that these effects were due to interference with peripheral retinal control of eye growth or destruction of hyalocytes with elimination of their synthesis of vitreous macromolecules, particularly the structural component hyaluronan (HA). Studies76 in humans who underwent peripheral retinal cryoablation for retinopathy of prematurity confirmed the finding of decreased eye growth in treated cases. Thus, because vitreous plays such an important role in eye growth, it is likely to have an important contribution in myopia. This could result either from primary abnormalities in the vitreous, or vitreous could play a secondary role mediating the effects of a primary abnormality either in the retina or ciliary body.
Vitreous Abnormalities Resulting in Myopia
In myopia, vitreous is liquefied and contains filaments with localized nodules.77 Biochemical studies78 in myopic humans found decreased collagen content and concentration in the central vitreous. Hexoamine concentration, an index of HA, was not only decreased in the central vitreous but in the posterior vitreous cortex as well. Confirmatory studies79 in experimental models of myopia found a decrease in vitreous protein concentration. Other studies80 determined that the decrease in protein concentration in the liquid vitreous was concurrent with an increase in the protein concentration of the gel vitreous. More recent studies81 of both the volumes of the gel and liquid compartments of vitreous as well as the protein content in these compartments found that although overall vitreous volume increased in experimental myopia, the volume of gel vitreous remained the same, and there was a marked increase in liquid vitreous volume. Comparing the liquid and gel vitreous of myopic eyes to controls with respect to protein content as well as the spectrum of proteins resolvable by polyacrylamide gel electrophoresis showed that whereas the protein profiles of the gel and liquid vitreous were similar on the day of hatching, they differed by day 14 after hatching. The investigators concluded that the accumulation of liquid vitreous in the myopic eye is not a dilution phenomenon, but rather the result of active synthesis of liquid vitreous. This is quite different from the degenerative liquefaction seen in aging in which the increase in liquid vitreous volume occurs in synchrony with a decrease in gel volume.60,82,83
Abundant synthesis of liquid vitreous could be the source of increased intraocular pressure, or perhaps more importantly, an increase in the pressure gradient differential between the vitreous body and the suprachoroidal space84 that results in elongation of the eye and myopia. Another important consideration is that because the vitreous in myopia is far more liquid than in an emmetropic eye, the different physical characteristics inherent to such a composition may interfere with the feedback regulatory signals that would normally modulate the process of vitreous synthesis and thereby limit the forces that influence growth of the eye. Such altered feedback regulation may be an important component in the pathogenesis of myopia.
In a retrospective, histopathologic study85 of 308 eyes with pathologic myopia, nonspecific degenerative changes of the vitreous were detected in 35.1%. These investigators likely were describing a prominence of liquid vitreous and likened this dystrophic finding to the degeneration seen in aging. Stirpe and Heimann86 found that in 10% of 496 highly myopic eyes, there was extensive vitreous liquefaction and condensation of the vitreous base, resulting in giant retinal tears in 70% of these patients. There were 87 eyes (17.5%) with a prominent posterior vitreous lacuna (i.e., a large pocket of liquid vitreous). These eyes had more prominent posterior staphylomas and a thin posterior vitreous cortex that was firmly adherent to the internal limiting lamina of the retina. Posterior retinal breaks, including macular holes, were present in 56% of these cases.
Retinal Abnormalities Mediated by Vitreous Resulting in Myopia
In 1985 Curtin87 revised Vogt’s ectodermal-mesodermal growth disparity theory to state that excess formation of vitreous by retina expands the sclera in a generalized fashion to produce myopia, or, in extreme cases, in a focal pattern to produce a posterior staphyloma. In this context, the primary abnormality is in the retina. A recent study88 found that non-subtype-specific nicotinic antagonists chlorisondamine and mecamylamine each inhibited the development of form-deprivation myopia. Moreover, dihydro-beta-crythroidine, a relatively selective competitive inhibitor for nicotinic receptors, inhibited mainly axial elongation. The investigators concluded that retinal or RPE nicotinic cholinergic receptors regulate postnatal ocular growth. In another experimental study89 an indirect cholinomimetic, diisopropylfluorophosphate (DFP) was demonstrated to reduce myopia by 58% in DFP-injected eyes rather than increasing myopia and elongation of the vitreous chamber as expected. The steady-state concentrations of acetylcholine and dopamine were increased by 54% and 36%, respectively, in eyes injected with DFP. As the combination of dopamine antagonists and DFP reduced myopia, the authors hypothesized that reduction is related to DFP effect on dopamine levels in the retina. Thus, primary abnormalities in retinal development could cause abnormal vitreous development, resulting in myopia, or deficient retinal metabolism in response to other factors could be the cause. For example, visual deprivation in monkeys90 induces myopia that is due to an increase in axial length of the eye.91 In humans, eyelid hemangiomas that occlude visual input were found to result in a 50% incidence of myopia.92
Ciliary Body Abnormalities Mediated by Vitreous Resulting in Myopia
Studies93 in rabbits have shown that vitreous glycoproteins are synthesized by the ciliary body. Recent studies94 in chick and human embryo eyes showed that both vitreous and internal limiting lamina components are synthesized by the ciliary body, but assembled farther posteriorly in the eye. Thus, there could be abnormalities in either the ciliary body synthesis or intravitreal assembly process that influence vitreous formation and internal swelling pressure with secondary effects on the size of the eye and the induction of myopia.
VITREOUS AGING AND PVD
Biochemical Changes
Using slit lamp biomicroscopy in a clinical setting, Busacca95 and Goldmann96 observed that after the ages of 45 to 50 years there is a decrease in the gel volume and an increase in the liquid volume of human vitreous. Eisner97 qualitatively confirmed these clinical findings in his postmortem studies of dissected human vitreous and observed that liquefaction begins in the central vitreous. In a large autopsy study of formalin-fixed human eyes, O’Malley98 provided quantitative evidence to support these observations. He found that more than half of the vitreous body was liquefied in 25% of persons age 40 to 49 and that this increased to 62% of those ages 80 to 89. Oksala99 used ultrasonography in vivo to detect echoes from gel-liquid interfaces in 444 normal human eyes and found progressive liquefaction with age. Vitreous liquefaction actually begins much earlier than the ages at which clinical examination or ultrasonography detects changes. Balazs and Flood100,101 found evidence of liquid vitreous after age 4 and observed that by the time the human eye reaches its adult size (ages 14 to 18) 10% to 12.5% of the total vitreous volume consists of liquid vitreous (Fig. 5). In these postmortem studies of 610 fresh, unfixed human eyes it was observed that after age 40 there is a steady increase in liquid vitreous that occurs simultaneously with a decrease in gel volume. By the ages of 80 to 90 years, more than half the vitreous body is liquid. The finding that the central posterior vitreous is where liquefaction begins100 and where fibers are first observed102,103 is consistent with the concept that dissolution of the hyaluronic acid (HA)-collagen complex results in the simultaneous formation of liquid vitreous and aggregation of collagen fibrils into bundles of parallel fibrils seen as large fibers.106 In the posterior vitreous such age-related changes often form large pockets of liquid vitreous.104
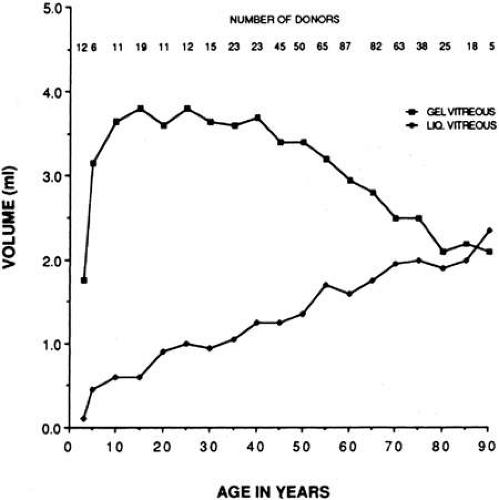 Fig. 5. Liquefaction of human vitreous. The volumes of gel and liquid vitreous in 610 human eyes were measured. The results are plotted versus the age of the donor. Liquid vitreous appears by the age of 5 and increases throughout life until it constitutes more than 50% of the volume of the vitreous during the tenth decade. Gel vitreous volume increases during the first decade while the eye is growing in size. The volume of gel vitreous then remains stable until about age 40, when it begins to decrease simultaneous with the increase in liquid vitreous.104 |
Age-related rheologic changes in the vitreous may result from an alteration in HA-collagen interaction. Chakrabarti and Park105 claimed that the interaction between collagen and HA is dependent on the conformational state of each macromolecule and that a change in the conformation of HA molecules could result in vitreous liquefaction and aggregation or cross-linking of collagen molecules. Armand and Chakrabarti106 have detected differences in the structure of the HA molecules present in gel vitreous and those in liquid vitreous, suggesting that such conformational changes occurred during liquefaction. Whether these changes are cause or effect is not known. However, Andley and Chapman107 have demonstrated that singlet oxygen can induce conformational changes in the tertiary structure of HA molecules. Ueno et al.51 have suggested that free radicals generated by metabolic and photosensitized reactions could alter HA and/or collagen structure and trigger a dissociation of collagen and HA molecules ultimately leading to liquefaction. This is plausible since the cumulative effects of a lifetime of daily exposure to light may influence the structure and interaction of collagen and HA molecules by the proposed free radical mechanism(s). The importance of vitreous liquefaction in the pathogenesis of PVD is discussed below.
Total vitreous collagen content does not change after the ages of 20 to 30 (Fig. 6). However, the collagen concentration in the gel vitreous at the ages of 70 to 90 (approximately 0.1 mg/mL) was found to be significantly greater than at the ages of 50 to 60 (slightly more than 0.05 mg/mL; P < 0.05). Since the total collagen content does not change, this finding is most likely due to the decrease in the volume of gel vitreous that occurs with aging (Fig. 5), and a consequent increase in the concentration of the collagen remaining in the gel. The collagen fibrils in this gel become packed into bundles of parallel fibrils,106 perhaps with cross-links between them. Aging of collagen throughout the body is associated with increased cross-linking as manifested by decreased solubility,108 increased collagen “stiffness,”109 and increased resistance to enzymatic degradation.110 There is also an increase in a reducible lysine-carbohydrate condensation product with increasing age.111 Although similar investigations have not been performed on vitreous collagen, studies by Snowden et al.112 demonstrated that with aging there is a decrease in the quantity of bovine vitreous collagen solubilized by heat alone. This could result from an increase in collagen cross-linking, or, as suggested by the authors, from changes in the surrounding glycoproteins and proteoglycans. Abnormal collagen cross-links have been identified in diabetic human vitreous113 suggesting “precocious senescence” of vitreous collagen as has been described in diabetes for other organs and tissues.114,115
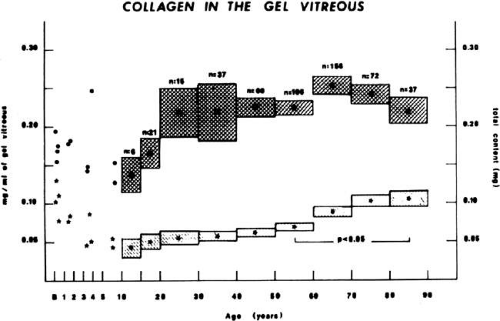 Fig. 6. Age-related changes in human vitreous collagen levels. Collagen content (mg; right ordinate) is indicated by solid dots (means) and darkly hatched boxes in the upper portion of the graph (height = standard error). Collagen concentration (mg/mL; left ordinate) is indicated by asterisks (means) and lightly hatched boxes in the lower portion of the graph (height = standard error). Vitreous collagen concentration decreases during the first decade of life because there is no net synthesis of collagen during this period of active growth of the eye. There are no significant changes in collagen content following age 20, consistent with the lifelong “stability” of this molecule. However, collagen concentration in the gel vitreous increases after ages 40 to 50. This is due to the decrease in gel vitreous that occurs during this time (Fig. 5), concentrating the remaining collagen in an ever-decreasing volume of gel vitreous. The increase in gel vitreous collagen concentration between ages 50 to 60 and 80 to 90 is statistically significant (P < 0.05).104 |
Vitreous HA concentration increases until about the age of 20, when adult levels are attained (Fig. 7A). HA concentration does not change in either the liquid or gel vitreous from ages 20 to 60 (Fig. 7).104,116 This necessarily means that there is an increase in the HA content of liquid vitreous and a concomitant decrease in the HA content of gel vitreous, since the amount of liquid vitreous increases and the amount of gel vitreous decreases with age (Fig. 2). This is consistent with the concept that vitreous liquefaction is associated with a “redistribution” of HA from the gel to the liquid vitreous. With advanced liquefaction there is a marked increase in the concentration of HA in liquid vitreous at ages 70 to 90 (Fig. 4B).
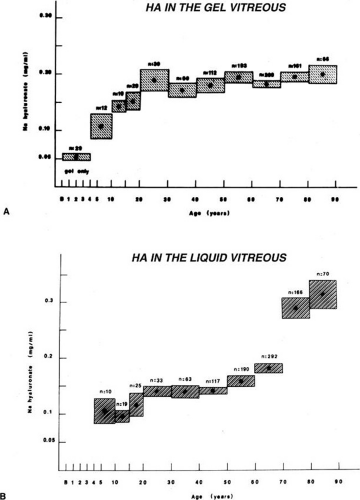 Fig. 7. Age-related changes in human vitreous hyaluronic acid (HA) concentration. The dots represent the means of the samples. The vertical height of the boxes represents the standard error of the means. The horizontal width of the boxes represents the age ranges in the sample group. A: HA concentration in the gel vitreous. There is a fourfold increase in the concentration of HA during the first two decades of life. Considering that this is also a period of active growth of the eye and substantial increase in vitreous volume, there must be prolific synthesis of HA to increase concentrations so dramatically. After age 20, HA concentrations in the gel vitreous are stable. Since this is a period of decreasing amounts of gel vitreous (Fig. 5), there must be a net decrease in the HA content of the gel to result in no substantial changes in concentration. B: HA concentration in the liquid vitreous. There are no data for the first 4 years since there is no liquid vitreous during this time. From the ages of 5 to 50 to 60 there is a 50% increase in the concentration of HA in the liquid vitreous. After this time there is a substantial increase in liquid vitreous HA concentration. The magnitude of this accumulation of HA in the liquid component of the vitreous is even greater when one considers that this occurs during the time when the volume of liquid vitreous is increasing by twofold (Fig. 5).104 |
Vitreous soluble protein concentrations also increase from 0.5 to 0.6 mg/mL at ages 13 to 50, to 0.7 to 0.9 mg/mL at ages 50 to 80, and 1.0 mg/mL above the age of 80.103,104,121 This increase may result from an age-related breakdown in the blood-ocular barriers of the retinal vasculature, retinal pigment epithelium, and ciliary body epithelium.
Structural Changes
Basal Laminae and Vitreous Base
The vitreoretinal interface is the site of many blinding vitreoretinal disorders.117 The basal laminae surrounding the vitreous thicken with age,118 a phenomenon that occurs in basal laminae throughout the body.119 Hogan et al.120 claimed that the thickening of the internal limiting lamina of the retina occurs during life as a result of synthesis by subjacent Müller’s cells. This phenomenon may play a role in weakening vitreoretinal adhesion, thus contributing to the development of PVD.
Teng and Chi121 found that the vitreous base posterior to the ora serrata varies in width depending on the age of the person. More than half of eyes from people older than age 70 had a posterior vitreous base wider than 1 mm. The width increased with advancing age to nearly 3 mm, bringing the posterior border of the vitreous base closer to the equator. This posterior migration of the vitreous base probably plays an important role in the pathogenesis of peripheral retinal breaks and rhegmatogenous retinal detachment since this is the area of strongest vitreoretinal adhesion. The phenomenon of posterior migration of the posterior border of the vitreous base has recently been confirmed and the increased vitreoretinal adhesion in this location has been hypothesized as due to intraretinal synthesis of collagen fibrils that penetrate the internal limiting lamina of the retina and “splice” with vitreous collagen fibrils.122 Within the vitreous base, Gartner123 found no differences in the thickness of collagen fibrils when comparing five eyes from humans ages 9 months and 29, 39, 61, and 71 years. He did, however, note that there was “lateral aggregation” of collagen fibrils in the eyes from older persons, similar to aging changes within the central vitreous.105,106,107 All these aging changes at the vitreous base very likely contribute to increased traction on the peripheral retina and the development of retinal tears and detachment.
Central Vitreous
During postnatal development there is a transition from a dense, highly light-scattering structure (Fig. 8) to homogeneous transparency (Fig. 9). The redistribution of HA and collagen during maturation to the adult results in the aggregation of collagen fibrils into packed bundles of parallel fibrils that appear as fibers. During the latter decades of life these fine parallel fibers in the central vitreous become thickened and tortuous (Fig. 10). Immediately adjacent to these coarse structures are areas with little or no light-scattering properties that are filled with “liquid” vitreous. When advanced, this vitreous degeneration forms large pools of liquid vitreous identified clinically as “lacunae” (Fig. 11). When the posterior vitreous detaches from the retina there is an overall reduction in the size of the vitreous body due to the collapse (syneresis) that occurs when liquid vitreous enters the retrohyaloid space posterior to the vitreous cortex and anterior to the retina This displacement of liquid vitreous occurs by way of the prepapillary “hole,” and possibly the premacular vitreous cortex, and is important in the pathogenesis of PVD.
 Fig. 8. Vitreous structure in a human embryo at 33 weeks of gestation. The posterior aspect of the lens is seen below. The vitreous body is enclosed by the dense, highly light-scattering cortex. Parapapillary glial tissue was torn away during dissection and hangs from the prepapillary vitreous cortex. Within the vitreous body, Cloquet’s canal arcs from the prepapillary vitreous toward the lens. Since its course undulates through the vitreous body, not all of Cloquet’s canal can be visualized in a single horizontal section.107 (Specimens courtesy of the New England Eye Bank, Boston.) |
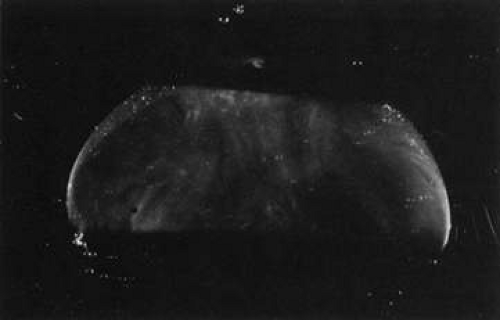 Fig. 9. Human vitreous structure in childhood. This view of the posterior and central vitreous from a 6-year-old child demonstrates a dense vitreous cortex with hyalocytes. There is vitreous extrusion into the retrohyaloid space through the premacular vitreous cortex. However, no fibers or other visible are present within the vitreous body.60,107 |
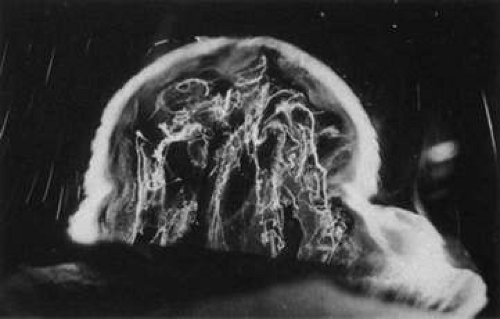 Fig. 10. Fibrous structure of human vitreous in old age. The vitreous body of an 88-year-old woman has undergone substantial degeneration in the fibrous structure. Fibers are thickened and tortuous. The entire vitreous body appears to have undergone dissolution with empty spaces adjacent to the thickened fibers.107 (Specimen courtesy of the New England Eye Bank, Boston.) |
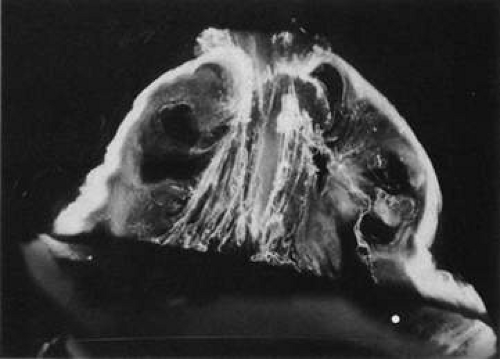 Fig. 11. Human vitreous lacunae in old age. The central vitreous has thickened, tortuous fibers. The peripheral vitreous has pockets devoid of any structure. These contain liquid vitreous and correspond to lacunae as seen clinically on biomicroscopy.107 |
Posterior Vitreous Detachment
The most common age-related event in the vitreous is PVD (Fig.12). True PVD can be defined as a separation between the posterior vitreous cortex and the internal limiting lamina (ILL) of the retina.124 PVD can be localized, partial, or total (up to the posterior border of the vitreous base). PVD should be distinguished from other forms of vitreoretinal separation that clinically may be mistaken for PVD. One of these forms involves separation of the ILL and some of the inner retina along with the detached posterior vitreous cortex. This can follow severe tractional events in the young, in whom the posterior vitreous cortex/ILL adhesion is strong.125 Lindner126 and Jaffe127 have described that in some cases of PVD there is herniation of vitreous through the vitreous cortex of the posterior pole. As previously mentioned, Gartner65 has drawn an analogy between this phenomenon and the herniation of the nucleus pulposus in the intervertebral disks of the spine. When a PVD involves herniation of the vitreous into the retrovitreal space by way of the premacular vitreous cortex, there can be persistent attachment to the macula and traction (Fig. 3).128
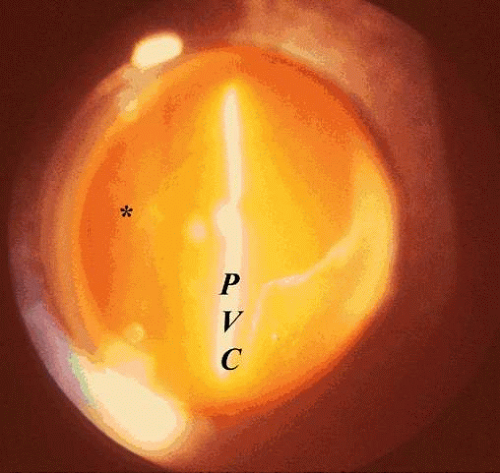 Fig. 12. Posterior vitreous detachment. Vitreous liquefaction (synchisis) in conjunction with dehiscence at the vitreoretinal interface allows liquid vitreous to dissect the posterior vitreous cortex (PVC) away from the internal limiting lamina of the retina and optic disc (*). Collapse (syneresis) of the vitreous anteriorly can be seen in the upper right corner of this clinical photograph taken by preset lens biomicroscopy. (Courtesy of C. L. Trempe, M.D.) |
Epidemiology of PVD
In clinical studies the incidence of purported PVD was found to be 53% in persons older than age 50, and 65% in those over age 65.131 Autopsy studies revealed an incidence of 27% to 51% in the seventh decade and 63% in the eighth decade.129 However, these figures may be overestimates due to the methods employed in these postmortem studies. PVD is more common in myopia, occurring 10 years earlier than in emmetropia and hyperopia.131 This likely results from effects of myopia on the structure of the vitreous. Cataract extraction in myopic patients introduces additional effects, causing PVD to develop in all but one of 103 myopic (greater than -6 D) eyes that were studied following cataract extraction (presumably intracapsular).130 Several studies131 have found a higher incidence of PVD in women than in men, a finding that may be due to hormonal changes following menopause. This hypothesis is supported by studies in the vitreous132 as well as other organs and tissues.133,134 A recent observational prospective clinical study135 found that following a PVD in one eye, the incidence of PVD in the fellow eye was 8% during the first 6 months, 24% within the first year, 80% within two years, and 90% after three years.
Pathogenesis of PVD
PVD results from rheologic changes within the vitreous that lead to synchisis (liquefaction), in conjunction with weakening of the vitreous cortex/ ILL adhesion. O’Malley101 suggested that PVD was strongly correlated with synchisis since both are correlated with age, although PVD had a later onset than synchisis. FOS and Wheeler134 studied 4,492 autopsy eyes and found a statistically significant correlation between the degree of synchisis and the incidence of PVD. Larsson and Osterlin136 studied 61 human eyes postmortem and found a positive correlation between the degree of vitreous liquefaction with the extent of PVD.
Once “liquid” vitreous forms and the collagen network is destabilized by the loss of HA molecules that strut and brace the collagen network, collapse (syneresis) of the vitreous body ensues. It is likely that dissolution of the posterior vitreous cortex/ILL adhesion at the posterior pole allows liquid vitreous to enter the retrocortical space by way of the prepapillary hole and perhaps the premacular vitreous cortex as well (Fig. 3).131,137 With rotational eye movements, liquid vitreous can dissect a plane between the posterior vitreous cortex and the ILL leading to true PVD. This volume displacement from the central vitreous to the preretinal space causes the observed collapse of the vitreous body (syneresis). It has long been recognized134,142 that the overwhelming majority of innocuous true PVD begins at the posterior pole. Vitreoretinal dehiscence at the macula may result from a predisposition or an increased stimulus for vitreous degeneration in the premacular region. Indeed, it is here that studies108 have identified a large pocket of liquid vitreous related to aging.138 Foos and Wheeler134 proposed that liquefaction in the posterior pole results from light toxicity to the premacular vitreous, since this is where the eye focuses incident light. There can also be a contribution of toxicity caused by metabolic waste products resulting from the high density of metabolically active neurons in the macula. Both light irradiation and metabolic processes can generate free radicals, which could alter HA and/or collagen structure and disrupt the HA-collagen association, causing liquefaction.51 These phenomena could also influence the extracellular matrix components binding the vitreous cortex to the ILL, contributing to the pathogenesis of PVD.
Sequelae of PVD
In youth the vitreous is transparent (Fig. 9) and has little or no effect on glare sensitivity.139 In old age, the aggregation of vitreous collagen fibrils into thick, irregular, visible fibers (Fig. 10) can induce glare sensitivity, which may be subjectively bothersome. Furthermore, the high incidence of PVD in old age may also induce glare due to scattering of light by the dense collagen fibril network in the posterior vitreous cortex. One group of individuals in whom glare discomfort is a common complaint are patients who have undergone scleral buckle surgery for rhegmatogenous retinal detachment. The complaint of glare appears to be due to postoperative vitreous turbidity and not a change in the threshold sensitivity of retinal receptors.140 Since vitreous biochemical and structural changes likely predispose these patients to rhegmatogenous retinal detachment, prominent vitreous changes are probably already present preoperatively. Scleral buckle surgery adds to the pre-existing vitreous inhomogeneity by breaking down the normal blood-vitreous barriers and mechanisms that maintain vitreous clarity.60
“Floaters” are the most common complaint of patients with PVD. These usually result from entoptic phenomena caused by condensed vitreous fibers, glial tissue of epipapillary origin that adheres to the posterior vitreous cortex and/or intravitreal blood.141 Floaters move with vitreous displacement during eye movement and scatter incident light, casting a shadow on the retina that is perceived as a gray, “hairlike” or “flylike” structure. Although not considered a disease in the classical sense, this condition can be extremely bothersome, prompting some patients to undergo vitrectomy. While this form of treatment is considered by most too aggressive for this problem, this may no longer be so, due to the advent of small (23 or 25 gauge) vitrectomy instruments. It is conceivable that the future will see the injection of pharmacologic agents to dissolve vitreous opacities and eliminate this problem even more safely.142,143 In an autopsy series of 320 cases with complete PVD, 57% had glial tissue on the posterior vitreous cortex.144 Vogt’s or Weiss’s ring are the terms applied when peripapillary tissue is torn away during PVD and forms a ring around the prepapillary hole in the posterior vitreous cortex. Murakami and et al.145 studied 148 cases of floaters and detected glial tissue on the posterior vitreous cortex in 83%. They claimed that patients complaining of multiple small floaters usually have minimal vitreous hemorrhage, frequently associated with retinal tears. Lindner131 found that minimal vitreous hemorrhage occurred in 13% to 19% of cases with PVD.
In 1935, Moore146 described that “light flashes” are sometimes a complaint resulting from PVD. Wise147 has noted that light flashes occurred in 50% of cases at the time of PVD and were usually vertical and temporally located. These are generally thought to result from vitreoretinal traction and thus are considered by most to signify a higher risk of retinal tears. However, Voerhoeff148 suggested that the light flashes are actually due to the detached vitreous cortex impacting on the retina during eye movement. It has been my personal observation that temporal light flashes that are arc-shaped and can be triggered by ocular saccades are commonly associated with retinal tears. Once PVD occurs there are differences in the characteristics of the detached vitreous which influence, for example, vitreous movement following displacement by ocular saccades. Differences in the “stiffness” of the detached vitreous have been identified using ultrasonic Doppler techniques.149 These probably relate to the biochemical composition and organization of the vitreous and are likely to change with time, depending on the effects of vitreous detachment on retinal and vitreous metabolisms.
ANOMALOUS PVD
In 1904, Best150 emphasized that every movement of the eye results in movement of the vitreous body and that this causes traction at any point of strong vitreoretinal adhesion. When vitreous liquefaction develops concurrently and in tandem with weakening at the vitreoretinal interface, an “innocuous” PVD results in clean separation of vitreous from retina, eliminating any further potential traction. Discordance between the rates and extents of these two processes results in “anomalous” PVD. This is most often due to advanced vitreous liquefaction without concomitant dehiscence at the vitreoretinal interface, which exerts traction upon the retina. The various manifestations of anomalous PVD differ depending upon where vitreous is most adherent to retina. The usual sites of strong adhesion are the vitreous base, optic disc, macula, over retinal vessels, and often a “sheetlike” adhesion in the posterior pole of young persons.130 In the periphery, anomalous PVD induces retinal tears and detachments. Vitreous adherence to blood vessels can result in retinal and/or vitreous hemorrhage following anomalous PVD. At the macula, anomalous PVD is important in vitreomacular traction syndrome, and via vitreoschisis can induce macular pucker and macular holes. At the optic disc, vitreo-papillary traction can induce visual disturbance151 and contribute to the pathogenesis of proliferative diabetic retinopathy. Anomalous PVD is thus a unifying concept that is a useful way to regard many vitreoretinal disorders that were previously perceived to be of entirely disparate natures.152 Table 1 lists how anomalous PVD causes various vitreoretinal pathologies, based upon the location of vitreous traction.
TABLE 1. Anomalous PVD | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Anomalous PVD and Peripheral Retinal Traction
Schepens153 first described the clinical appearance of peripheral vitreoretinal traction as “white with or without pressure,” the former referring to the appearance of the peripheral fundus when examined during scleral depression and the latter being the appearance without scleral depression. Daicker154 proposed that these appearances result from “collagenic” formations in the peripheral retina, while Gartner155 suggested that they are due to irregularities of the internal limiting lamina. Green and Sebag156 described that the appearance of these ophthalmoscopic findings results from incident light (from the ophthalmoscope) that is tangential to dense bundles of vitreous collagen. Watzke157 performed clinicopathologic correlation of a case with this finding and described that the lesion was due to portions of the vitreous cortex that remained attached to the retina following PVD. This might therefore represent a variant of vitreoschisis (splitting of the vitreous cortex), which in this instance occurs in the peripheral fundus. Some159 consider that “white with or without pressure” predisposes to peripheral retinal tears. However, Byer158 does not believe that this ophthalmoscopic appearance has any diagnostic or prognostic significance.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


