Chapter 21 Vitreous and Vitreoretinal Interface
Biochemistry of vitreous
Collagen
Collagen is the major structural protein, consisting of heterotypic fibrils (Fig. 21.1) similar to cartilage.1,2 Following vitrectomy, a type II procollagen is secreted in humans,3 but vitreous is not reformed, since reoperations reveal that the gel state of vitreous is not re-established.
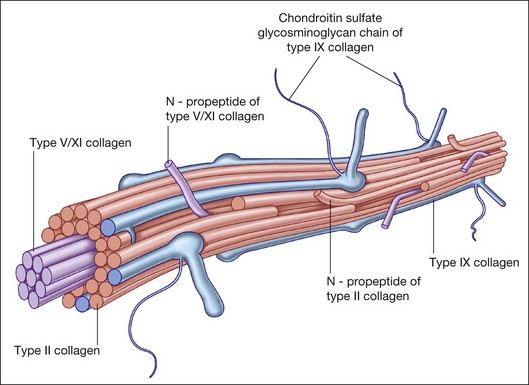
Type II collagen4 comprises 75% of the total collagen content in vitreous. There are considerable similarities between vitreous and cartilage collagens,5,6 perhaps explaining why inborn errors of type II collagen metabolism result in “arthro-ophthalmopathies,”7 manifesting similar phenotypic expression in joints and vitreous. Type IX collagen accounts for up to 15% of vitreous collagen,8 where it always contains a chondroitin sulfate glycosaminoglycan chain9 covalently linked to the α2 (IX) chain at the NC3 domain, enabling the molecule to assume a proteoglycan form.
An important function of vitreous is maintaining transparency within the eye (Fig. 21.2).2 Studies10 have shown that one of the minor collagens of vitreous is type XVIII, progenitor of endostatin, a potent inhibitor of angiogenesis.11

Hyaluronan
Hyaluronan (HA) was first isolated from bovine vitreous in 1937. HA appears after birth, perhaps synthesized by hyalocytes,2 the ciliary body, and/or Müller cells. It is a large polyanion, which can influence the diffusion of drugs through vitreous.12,13 As a result of HA’s entanglement within the vitreous collagen fibril matrix, the mechanical force of HA’s extension and contraction can be transmitted to the retina, optic disc, and neovascular complexes, inducing untoward effects in conditions with fluctuations in ionic balance and hydration, such as diabetes.14
Chondroitin sulfate
Most vitreous chondroitin sulfate is in the form of versican,15 believed to form complexes with HA as well as with microfibrillar proteins such as fibulin-1 and fibullin-2 and play a crucial role in maintaining the molecular morphology of vitreous.16 Mutations that alter the splicing of the central chondroitin sulfate-bearing domains of versican have been implicated in Wagner syndrome, a condition with excess vitreous liquefaction.17
Noncollagenous structural proteins
Fibrillins
In Marfan syndrome defects in the gene encoding fibrillin-1 (FBN1 on chromosome 15q21) result in both ectopia lentis and vitreous liquefaction,18 important in rhegmatogenous retinal detachment (RD) in these patients.
Opticin
A major noncollagenous protein of vitreous is opticin (formerly vitrican).19 It is bound to the surface of the heterotypic collagen fibrils and prevents aggregation of adjacent collagen fibrils into bundles. Opticin binds heparan and chondroitin sulfates, suggesting that opticin may play a role in vitreoretinal adhesion.20,21 Similar to its role in articular cartilage,22 opticin may also stabilize vitreous gel structure by binding chondroitin sulfate proteoglycans.
Anatomy and histology
Vitreous body
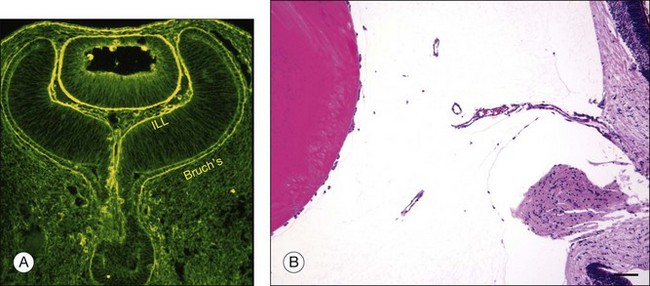
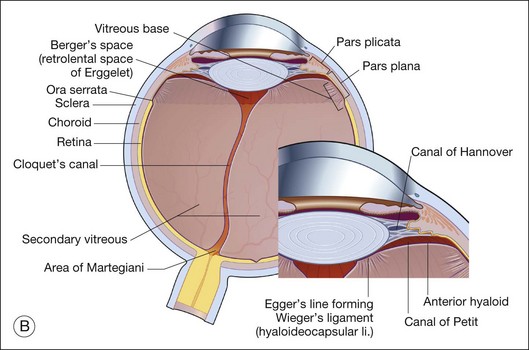
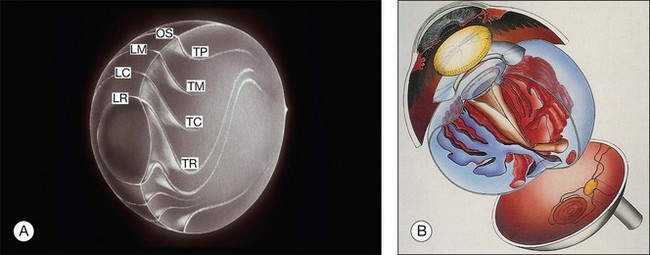
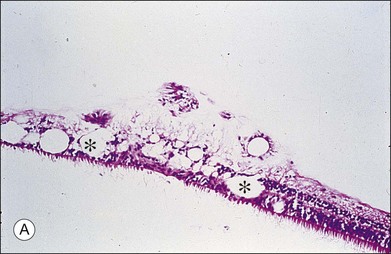
Vitreous is a clear gel-like structure with a volume of 4.0 mL. During invagination of the optic vesicle the “primary” vitreous forms between the lens and the internal limiting lamina (ILL on Fig. 21.3A) of the retina. It is noteworthy that the ILL is continuous with Bruch’s membrane, demonstrating a common embryologic origin with analogous molecular composition and structure, suggesting important similarities later in life.23 The “secondary” vitreous begins to develop at the 13-mm stage of embryogenesis and is derived from the retina and mesoderm of the hyaloid vascular system (Fig. 21.3B).
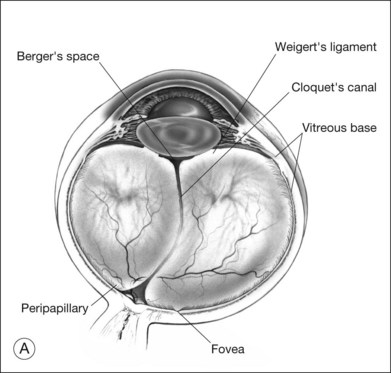
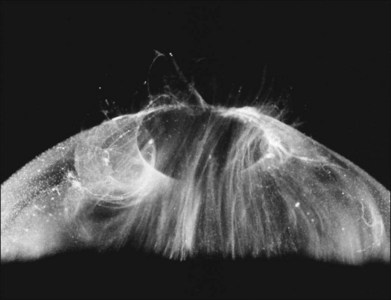
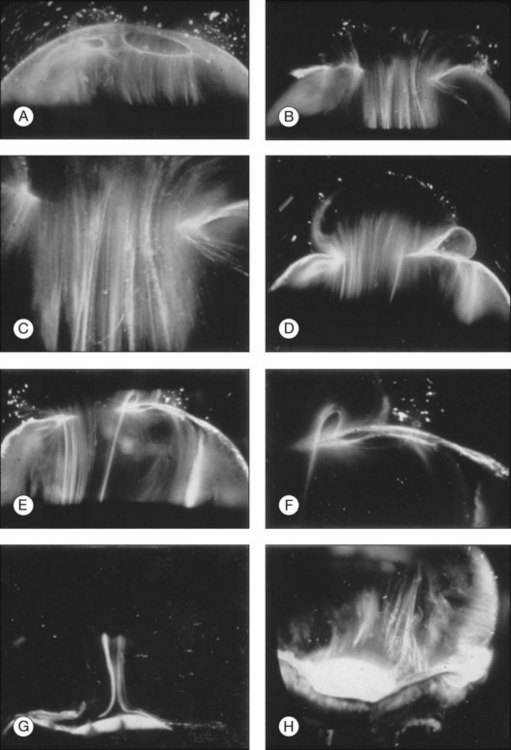
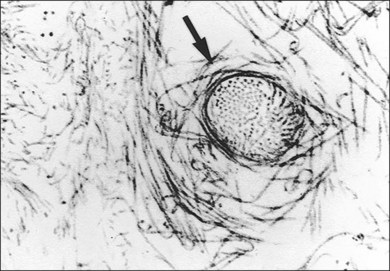
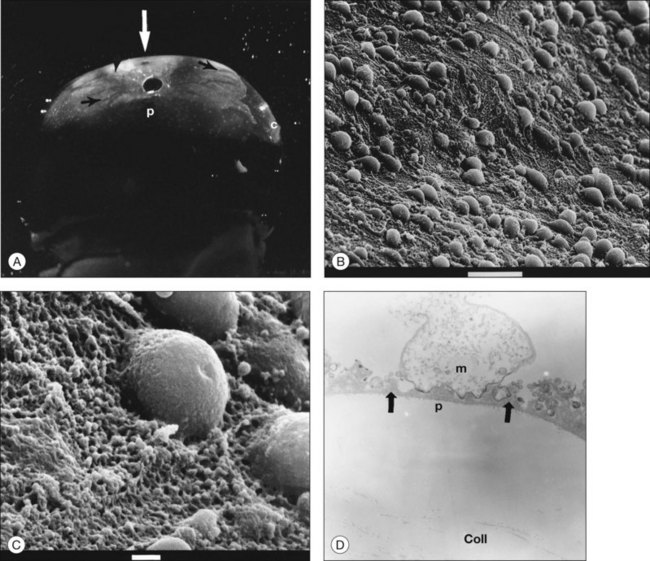
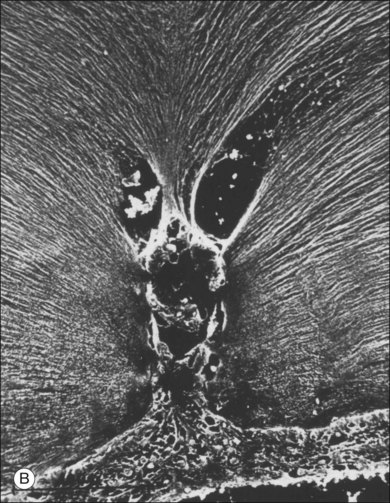
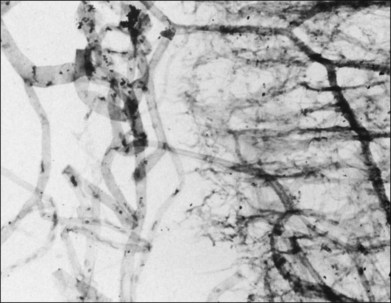
Classic depictions of human vitreous structure are shown in Fig. 21.4. Modern concepts proposed membranous (Fig. 21.5A)24 and cisternal (Fig. 21.5)25 systems. Sebag and Balazs26 performed dark-field slit microscopy to define the posterior vitreous cortex as a thin, membranous structure continuous from the ora serrata to the posterior pole. Two round holes are present in the prepapillary and premacular areas (Fig. 21.6). Anteroposterior fibers (Fig. 21.7) comprised of parallel collagen fibrils (Fig. 21.8) arise from the vitreous base (Fig. 21.9A), where Gartner27 found “lateral aggregation” in older individuals. Vitreous base collagen fibers insert anterior to the ora serrata forming the anterior loop (Fig. 21.9B), important in anterior proliferative vitreoretinopathy (PVR).28 In the posterior pole, fibers extend through the premacular hole (Figs 21.6 and 21.7A), but a few attach to the rim of the hole. Condensed bundles of fibers insert into the vitreous cortex in the midperiphery and equator (Fig. 21.10).


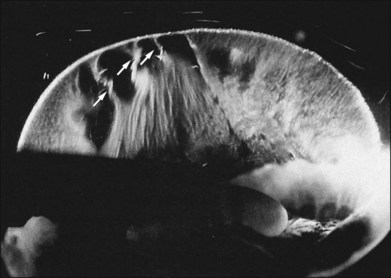
Vitreoretinal interface
Posterior vitreous cortex
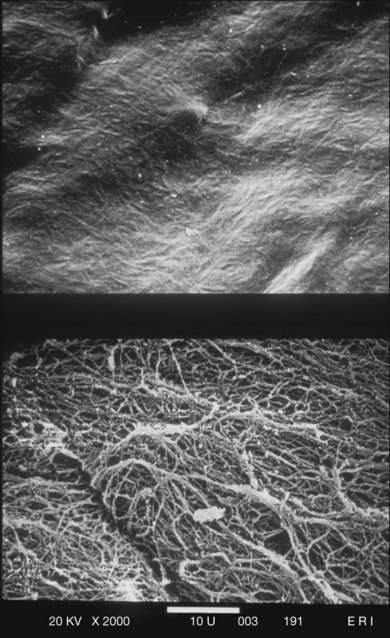
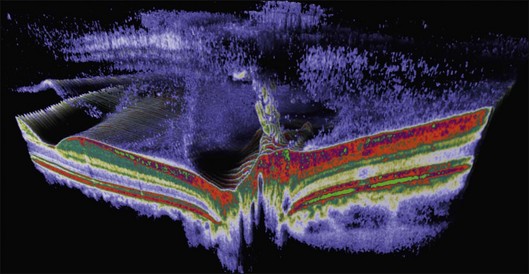
The posterior vitreous cortex is 100–110 µm thick and consists of densely packed collagen fibrils29 (Fig. 21.11). There is no vitreous cortex over the optic disc (Figs 21.6 and 21.7A), and the cortex is thin over the macula. The prepapillary hole can sometimes be visualized clinically following posterior vitreous detachment (PVD). If peripapillary tissue is torn away during PVD and remains attached around the prepapillary hole, it is called Vogt’s or Weiss’s ring. Gupta et al.30 demonstrated a lamellar organization of the posterior vitreous cortex (Fig. 21.12), confirmed in humans by three-dimensional optical coherence tomography (OCT) (Fig. 21.13).31 During anomalous PVD (APVD)32 these predispose to splitting along potential cleavage planes, resulting in vitreoschisis.33
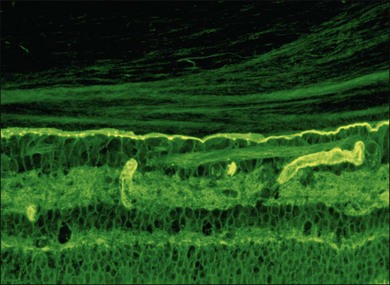
Hyalocytes
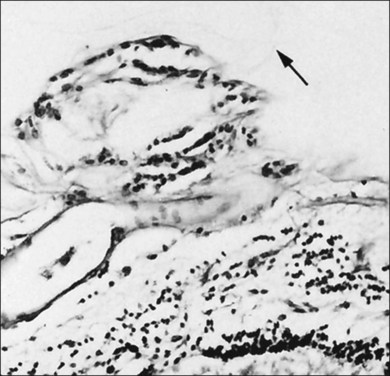
Hyalocytes are mononuclear cells embedded in the posterior vitreous cortex 20–50 µm from the ILL posteriorly (Figs 21.6 and 21.14). The highest density of hyalocytes is in the vitreous base followed by the posterior pole, with the lowest density at the equator.34,35 Balazs36 suggested that these cells synthesize vitreous HA,37–40 but Swann5 disagreed. Evidence suggests that hyalocytes maintain ongoing synthesis and metabolism of glycoproteins41,42 and may also synthesize collagen43 and enzymes.44
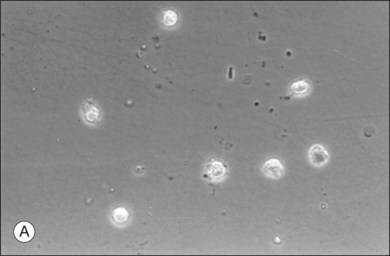
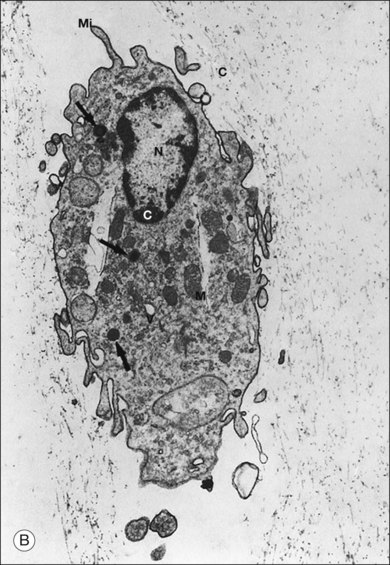
The phagocytic capacity of hyalocytes has been described in vivo45 and demonstrated in vitro.46–48 Hyalocytes become phagocytic in response to inducting stimuli and are important in antigen processing and as initiators of the immune response, making possible intravitreal inoculation of antigens to promote systemic immunity.49 HA may have a regulatory effect on hyalocyte phagocytic activity.50,51 Various constituents of vitreous52,53 may be immunogenic and play a role in ocular inflammatory diseases. Sakamoto and Ishibashi have recently published an excellent review of hyalocytes.54
Hyalocytes are important in macular pucker when APVD28 and vitreoschisis26,29 leave these cells on the macula (Fig. 21.15). Under the influence of cytokines, hyalocytes proliferate55 on the surface of the retina, resulting in hypercellular membranes. Hyalocytes also recruit cells from the circulation and the retina (glial cells) via the release of connective tissue growth factor and induce collagen gel contraction in response to platelet-derived growth factor and other cytokines,56,57 causing tangential vitreoretinal contraction.
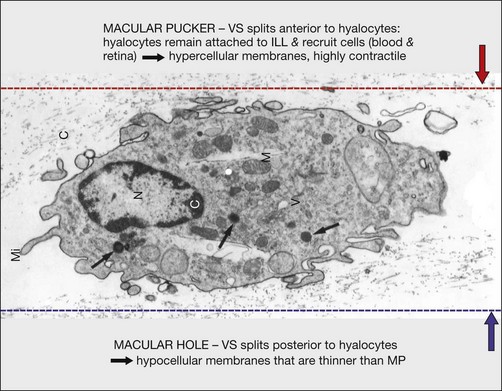
Fig. 21.15 Vitreoschisis (VS). Transmission electron microscopy of human hyalocyte (same as in Fig. 21.14B) demonstrating two potential levels splitting during vitreoschisis. Anomalous posterior vitreous detachment that induces vitreoschisis which splits the posterior vitreous cortex anterior to the level of hyalocytes (red dashes) leaves these cells attached to the macula, resulting in a hypercellular membrane. The lack of attachment to the optic disc allows inward (centripetal) tangential traction causing contraction and macular pucker. If vitreoschisis splits the posterior vitreous cortex posterior to the level of hyalocytes (blue dashes), the remaining membrane is thin and hypocellular. If there is also vitreopapillary attachment, the tangential forces will be outward (initially nasally), opening a central dehiscence and inducing a macular hole. ILL, internal limiting lamina; VS, vitreoschisis; MP, macular pucker; for other abbreviations see Fig. 21.14.
Internal limiting lamina (ILL) of the retina
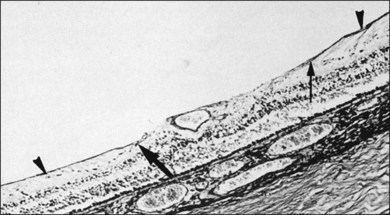
The ILL is a multilaminar structure of variable thickness topographically. Adjacent to Müller cell foot plates is the lamina rara externa (0.03–0.06 µm) with no species variations or changes with topography or age. The lamina densa is thinnest at the fovea (0.01–0.02 µm) and thicker in the posterior pole (0.5–3.2 µm) than at the equator or vitreous base, where Foos58 found the ILL to be uniformly thin (51 nm) and the lamina rara to be 40 nm wide with traversing fibrils that were denser at sites corresponding to attachment plaques in Müller cells. The ILL is very thin over major retinal vessels (Fig. 21.16) where defects allow glial cells to extend on to the inner retina.59 Acquired ILL defects are in the foveola, retinal pits, retinal tufts, and retinal lattice. The ILL progressively thickens posteriorly to about 306 nm at the equator and about 1887 nm posteriorly. Müller footplates are less numerous at the equator than at the vitreous base. Posteriorly, no Müller footplates were observed and the inner aspect of the ILL remains smooth, while the outer aspect is irregular. Peripherally, both inner and outer surfaces are smooth.2,53
The ILL consists of type IV collagen, associated with glycoproteins,23,60,61 type VI collagen, which may contribute to vitreoretinal adhesion, and type XVIII,62 which binds opticin.63 Opticin binds to heparan sulfate, contributing to vitreoretinal adhesion.64 Type XVIII collagen also prevents cell migration from the retina into vitreous.65
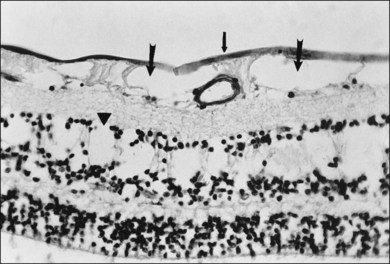
Retinal sheen dystrophy66
This ILL dystrophy has cystic spaces under the ILL and in the inner nuclear layer (Fig. 21.17), and numerous areas of separation of the ILL from the retina with filamentous material (Fig. 21.18). Endothelial cell swelling and degeneration, pericyte degeneration, and basement membrane thickening of retinal capillaries suggest that this condition is primarily a retinal vasculopathy with edema, swelling, and degeneration of Müller cells. Alternatively, the primary defect could be in Müller cells.
Degenerative remodeling
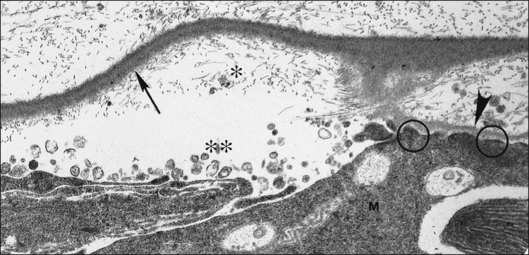
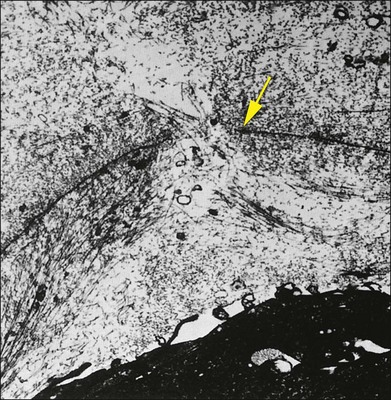
Foos67 defined a spectrum of changes in the ILL as “degenerative remodeling.” Features include detachment and discontinuity of the ILL with vitreous collagen beneath the ILL (Fig. 21.19), cellular debris with macrophages, and absence of Müller cell attachment plaques. In larger lesions, vitreous may insinuate into degenerative crypts and adhere to the cell membrane of the lining Müller cells that have no basal lamina (Fig. 21.20). In the peripapillary area, retinal glial cells extend from the optic disc and are continuous with a glial epipapillary membrane that has vitreous fiber incarceration. Roth and Foos68 observed nasal epipapillary membranes associated with Bergmeister papillae in 27.6% of autopsy eyes.
Vitreoretinal interface
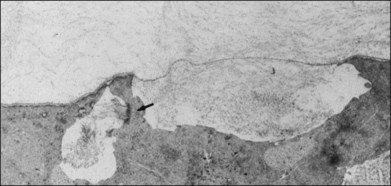
The interface between vitreous and adjacent structures consists of a complex formed by the vitreous cortex and basal laminae which are firmly attached to their cells.69 The only part of vitreous not adjacent to a basal lamina is the annulus of the anterior vitreous cortex, which is directly exposed to the zonules and the aqueous humor of the posterior chamber, similar to the surface of articular cartilage, which is exposed to synovial fluid.48 Zimmerman and Straatsma70 claimed that there are fibrillar attachments between the posterior vitreous cortex and the ILL. The composition of these fibrillar structures is not known and their presence has never been confirmed.
It is currently believed that an extracellular matrix “glue” of fibronectin, laminin, and other extracellular matrix components71 exists between vitreous and retina, causing adhesion to be fascial, as opposed to focal.55,56 Chondroitin sulfate is present at the sites of strong vitreoretinal adhesion such as the vitreous base and optic disc, forming the rationale for pharmacologic vitreolysis using avidin–biotin complex chondroitinase.
Topographic variations
Strength of vitreoretinal adhesion
Vitreous is attached to all contiguous structures, but is most firmly adherent at the vitreous base,72 which includes the posterior 2 mm of the pars plana and extends 1–4 mm posterior to the ora serrata, varying with age73 and topography (more posterior temporally). There are also topographic differences posteriorly, with greater adhesion at the posterior pole than the equator60 (Fig. 21.21).
Peripheral fundus and vitreous base
The vitreous base is a three-dimensional structure that straddles the ora serrata. There is a high density of collagen fibrils oriented at right angles to the inner surface of the ciliary epithelium and peripheral retina. The fibrils attach to the basement membrane of the nonpigmented epithelium of the pars plana and the ILL of the peripheral retina,74 intimately interwoven with an intervening extracellular matrix. Within the vitreous base, there are several anatomic variations where vitreoretinal adhesion is firm75 and associated with retinal breaks76,77:
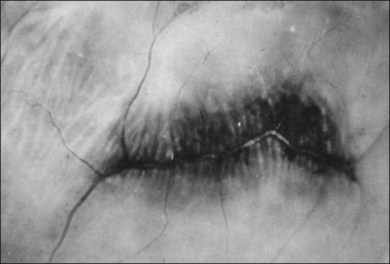
1. Ora bays70,75,78 (Fig. 21.22)
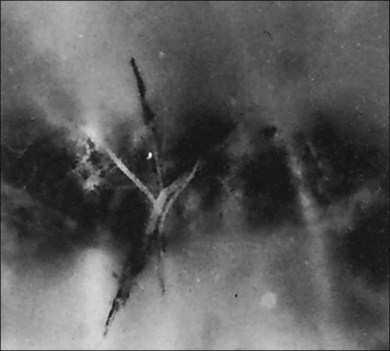
2. Meridional folds77,79 (Fig. 21.23)
3. Meridional complexes77 (Figs 21.22 and 21.24)
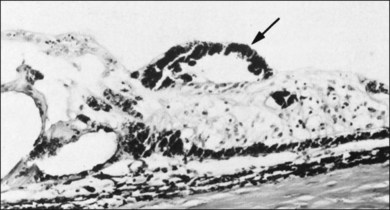
4. Peripheral retinal excavations77,80 (Fig. 21.24)
6. Spiculate and nodular pigment epithelial hyperplasia87 (Fig. 21.28)
7. Retinal lattice “degeneration”58,84,89–105 (Figs 21.29–21.33)
8. White-with-pressure, white-without-pressure106–113
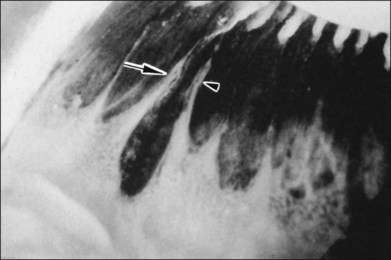
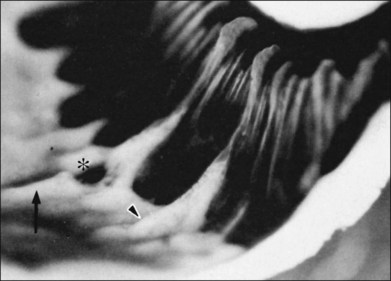
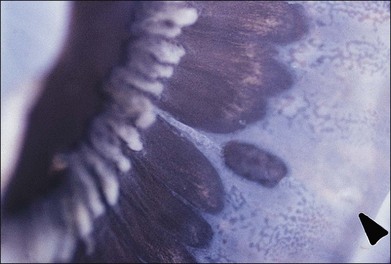
Fig. 21.24 Peripheral retinal excavation (arrowhead) in line with a meridional complex and an enclosed ora bay.
(Reproduced with permission from Green WR. Pathology of the retina. In: Frayer WC, editor. Lancaster course in ophthalmic histopathology, unit 9. Philadelphia: FA Davis; 1981.)
Interface along major retinal vessels
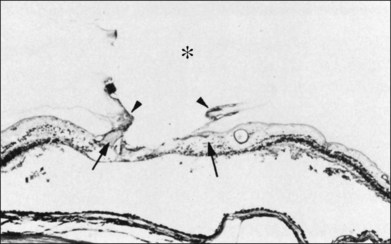
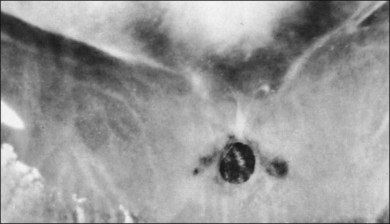
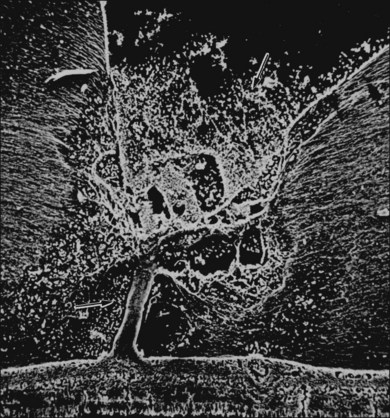
The ILL thins and is sometimes absent over major retinal vessels115,116 (Fig. 21.35). At such points, vitreous may be incarcerated into retina, directly continuous with perivascular tissue, attachments called “vitreoretinovascular bands,”117 or “spider-like bodies,”118 purported to be vitreous fibrils that traverse the ILL and coil about retinal blood vessels. It is common for retinal vessels to be associated with paravascular rarefaction (cystic degeneration), retinal pits and tears, and avulsion of retinal vessels.
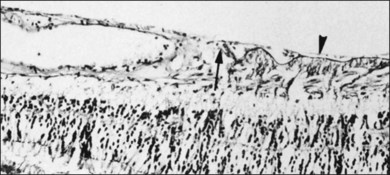
Vitreomacular interface
Attachment of vitreous to the macula occurs in an irregular, annular zone of 3–4 mm diameter,2 generally not visible by clinical examination in normal adults but possibly evident in fetal and young adult eyes and in pathologic conditions. The posterior vitreous cortex is thinner over the macula in a disc-shaped area about 5 mm in diameter. Discontinuity of the ILL in the fovea may be a site where glial cells extend on to the inner surface of the retina (Fig. 21.36).
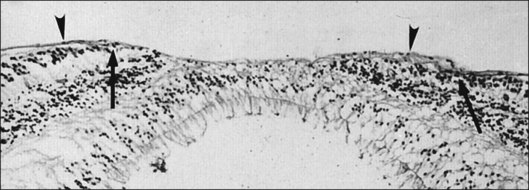
Vitreopapillary interface
At the rim of the optic disc the ILL ceases, although the basement membrane continues as the inner limiting membrane of Elschnig.119 This membrane is 50 nm thick and is believed to be the basal lamina of the astroglia in the optic nerve head. At the centralmost portion of the optic disc the membrane thins to 20 nm, follows the irregularities of the underlying cells of the optic disc, and is composed only of glycosaminoglycans and no collagen (central meniscus of Kuhnt). Given that the ILL prevents the passage of cells,47 the thinness and chemical composition of the central meniscus of Kuhnt and the membrane of Elschnig may account for frequent cell proliferation from or near the optic disc.
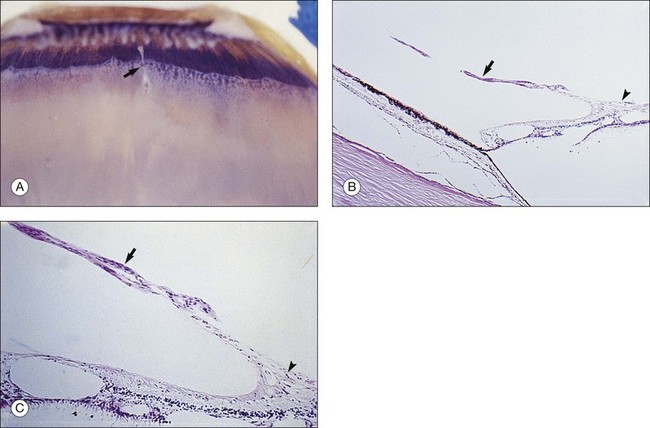
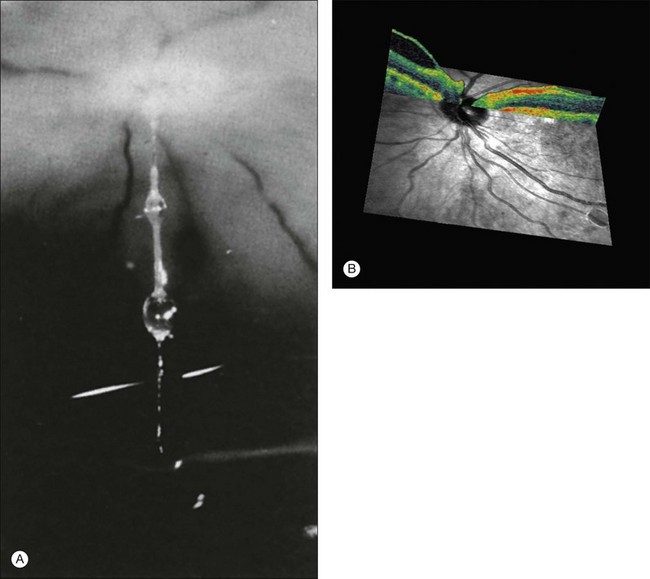
Vitreous attachment to the optic disc may persist even though the vitreous is detached elsewhere120 (Fig. 21.37). This adhesion may be fortified by epipapillary membranes.68 The entire complex may subsequently detach, resulting in a ring of tissue composed of fibrous astrocytes and collagen (Fig. 21.38) that flutters in and out of the visual axis, causing “floaters.” Vitreopapillary adhesion (VPA) contributes to macular holes121 and any vitreomaculopathy that features intraretinal cystoid spaces,122 presumably due to the influence upon the vectors of tangential traction upon the macula.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


