Vascular Anomalies of the Fundus
Adam T. Gerstenblith
Jeremy D. Wolfe
Gary C. Brown
Choroidal and retinal vascular anomalies may be associated with systemic disorders. Accurate diagnosis is therefore critical. In most instances, careful ophthalmoscopic examination is sufficient; however in some cases, ancillary tests such as intravenous fluorescein angiography (IVFA), indocyanine green (ICG) angiography, optical coherence tomography (OCT), ultrasonography, computed tomography (CT) and magnetic resonance imaging (MRI) are necessary either to confirm the diagnosis or to rule out associated systemic disorders.
CHORIOVAGINAL VEIN
A choriovaginal vein is an uncommonly recognized, unusually large choroidal vessel located within the posterior pole. The choriovaginal vein passes from the choroid through the sclera at the margin of the optic disc to terminate in the venous plexus of the pial sheath of the optic nerve.1 This establishes an indirect communication between the choroidal and retinal venous systems through the pial branches of the central retinal vein.2 It is generally seen temporal to the optic nerve and is predominantly noted in lightly pigmented, myopic fundi1,3 (Fig. 22-1). This entity was described in a case of trisomy 13 with congenital glaucoma, although it is not typically associated with any systemic abnormalities.4
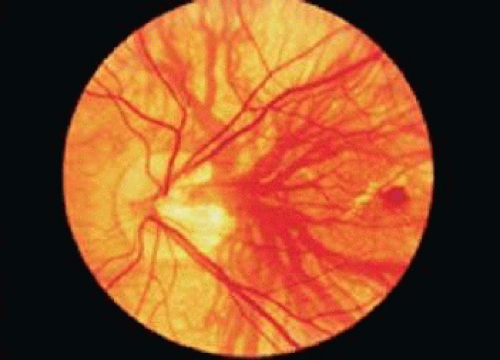 Figure 22-1. A choriovaginal vein (posterior vortex vein) situated just temporal to the optic disc in a highly myopic eye. These unusually located choroidal vessels are believed to be tributaries of the ophthalmic vein. A Fuchs spot is present in the fovea. |
PERSISTENT HYALOID ARTERY
The hyaloid vessels develop around the fifth week after conception. The hyaloid artery provides the bulk of blood supply to the developing posterior segment and lens during late embryologic and early fetal development. Its branches include the tunica vasculosa lentis and the vasa hyaloidea propria. Spontaneous regression of the fetal hyaloid artery commences at the start of the third trimester (28 weeks).5,6,7 Ultrasound examination during pregnancy typically demonstrates the hyaloid artery in fetuses of 20 weeks’ gestational age or younger, whereas its presence on ultrasound in the mid–third trimester is uncommon in healthy fetuses.8
A persistent hyaloid artery represents incomplete regression and is noted clinically as a single threadlike vessel emanating from the optic disc. It travels in a sinuous course anteriorly through the vitreous cavity within the Cloquet canal. Persistent hyaloid arteries can traverse the entire length of the vitreous cavity to insert on the posterior capsule of the lens (Fig. 22-2). When all but the anterior lens insertion of the hyaloid artery regresses, the remaining focal posterior lens capsule opacity is called the Mittendorf dot. At birth, up to 95% of premature infants have hyaloid artery remnants. Such remnants also have been noted in 3% of full-term infants.6 Persistent hyaloid arteries may be filled with blood. When they are blood filled, vitreous hemorrhage may occur. This can be spontaneous, precipitated by trauma or by a posterior vitreous detachment.9,10 It has been postulated that rapid eye movements during sleep produce traction, leading to rupture of a freely floating hyaloid artery in young individuals.11 Other ocular conditions that may be associated with a persistent hyaloid artery include strabismus, cataract, amblyopia, and nystagmus.10 A persistent hyaloid artery typically does not need treatment, but recurrent vitreous hemorrhage secondary to this condition may require vitrectomy. Also, a persistent, patent hyaloid artery may complicate attempts at surgical repair in infants with the condition called persistent hyperplastic primary vitreous or with retinal detachment caused by retinopathy of prematurity.12
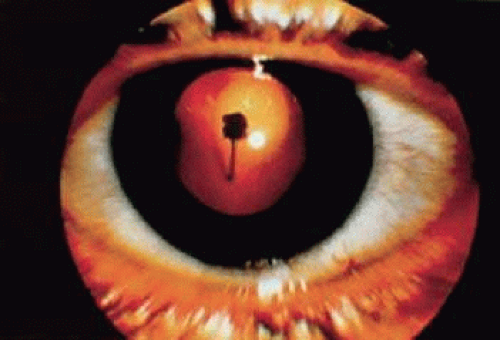 Figure 22-2. Slitlamp view of a persistent hyaloid artery inserting on the posterior lens capsule. |
PREPAPILLARY LOOPS
Prepapillary vascular loops were first recognized by Liebrich in 1871.13 These congenital vascular anomalies extend into the vitreous cavity. They appear as corkscrew, spiral, or hairpin vessels overlying the optic disc and are contiguous with the normal retinal circulation (Fig. 22-3A). They can arise from the central retinal vessels, a branch retinal vessel, or a cilioretinal vessel,6,7,14 About one third of prepapillary loops are encased in a fibroglial sheath, and they are present bilaterally in approximately 10% of affected persons.6 Eyes containing a prepapillary loop have an unusually high prevalence of associated cilioretinal arteries. On ophthalmoscopic examination, prepapillary loops may pulsate. IVFA has shown that 95% of prepapillary loops are arterial in origin.6,15
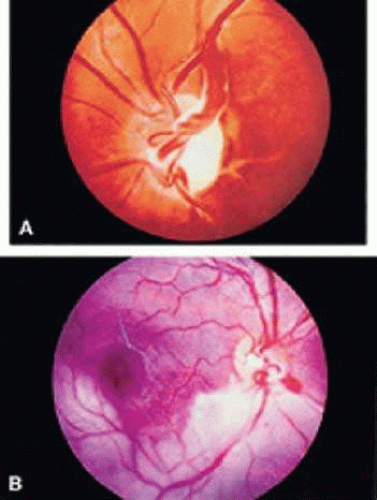 Figure 22-3. A. A prepapillary loop derived from the central retinal artery. Notice the fibrous sheath. B. Branch retinal artery occlusion associated with a prepapillary loop. |
Prepapillary loops were once believed to be remnants of the hyaloid artery, It is now thought that they represent developmental anomalies that occur when mesenchymal cells destined to differentiate into mature retinal vessels proliferate within the Bergmeister papilla.16 Histopathologic study has demonstrated communication of a prepapillary loop with the retinal arterial system.16 The prepapillary loop was supported by a connective tissue sheath that was lined by a cellular lamina continuous with the inner retina.
The major complication occurring with prepapillary loops is retinal arterial obstruction within the territory supplied by the loop (Fig. 22-3B). This is believed to occur secondary to turbulent flow, endothelial damage, and subsequent thrombus formation.15 Vitreous hemorrhage, amaurosis fugax, hyphema, and retinal venous macrovessel also have been reported.15,17,18,19,20 There are no associated systemic abnormalities. Two studies report a familial occurrence of prepapillary loops, although a definite hereditary pattern has not been identified.21,22
HEREDITARY RETINAL ARTERY TORTUOSITY
Hereditary or familial retinal artery tortuosity (fRAT) is a rare, congenital, autosomal dominantly inherited condition characterized by extreme tortuosity of the retinal arteries. Patients with this entity may present acutely with visual loss from a superficial macular hemorrhage that occurs spontaneously, after relatively minor trauma or physical exertion.14,23,24,25 Vision typically returns to normal as the retinal hemorrhage clears. The artery tortuosity is most marked in the macular region, whereas the retinal veins remain normal (Fig. 22-4). The vascular tortuosity progressively increases with age.25 On IVFA the tortuous retinal arteries are competent, and there is no microvascular cause for the macular hemorrhage. Histologic study by light microscopic examination in one case failed to reveal any structural abnormality in the arteriolar wall.23 No systemic vascular abnormality has been detected in these patients.26 One case report, however, of a 16-year-old boy with hereditary retinal artery tortuosity and recurrent macular hemorrhages found low levels of factor VII activity on coagulation studies. Although coagulation deficiencies are not frequently found in patients with fRAT, coagulation studies may be indicated, particularly in patients with recurrent hemorrhages.27,28 The other syndrome that typically produces isolated retinal arteriolar tortuosity without venous involvement is coarctation of the aorta.29 It may be reasonable to examine family members for arteriolar tortuosity before subjecting an affected individual to an extensive medical evaluation. Hereditary retinal artery tortuosity should be differentiated from other diseases that can induce retinal vascular tortuosity (affecting both the arteries and veins) including hypoxic retinal syndromes (e.g., sickle cell disease, congenital heart disease), carotid-cavernous sinus fistulas, leukemia, dysproteinemias, familial dysautonomia, mucopolysaccharidosis, and Fabry disease.14,30
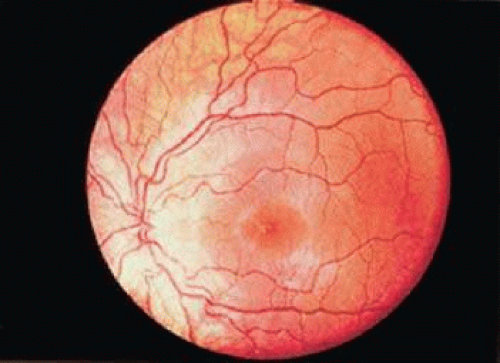 Figure 22-4. Hereditary retinal artery tortuosity in a 15-year-old asymptomatic boy, Notice the normal-appearing retinal veins. The patient’s father had a similar retinal vascular pattern. |
INHERITED RETINAL VENOUS BEADING
Meredith31 reported a family in whom some of the members presented with prominent bilateral beading and segmentation of their retinal veins. Associated findings included beading of the conjunctival veins, retinal capillary nonperfusion, microaneurysm formation, altered vascular permeability with lipid exudation, retinal edema, retinal neovascularization, and vitreous hemorrhage. The family’s pedigree was consistent with autosomal dominant inheritance. These retinal vascular changes may represent an unusual manifestation of renal disease, since two of the five patients concurrently had Alport syndrome. A second reported pedigree showed no evidence of renal disease, although affected members did display a relative neutropenia on peripheral blood examination.32
RETINAL MACROVESSELS
A congenital retinal macrovessel is an aberrant, enlarged branch retinal vessel that supplies or drains the macular area, crossing the horizontal raphe (Fig. 22-5A). They usually are veins. The condition is typically unilateral, benign, and ophthalmoscopically stable. This entity is often detected on routine examination.14 Visual acuity is typically unaffected, although concomitant foveal cysts have been noted.33 Rarely, serous retinal detachments may occur, particularly in the setting of sudden changes in gravitational forces.34,35 IVFA may reveal delayed drainage of dye from the macrovessel, associated microvascular abnormalities, and retinal capillary nonperfusion33 (Fig. 22-5B). On occasion, retinal macrovessels may be accompanied by similar vascular anomalies affecting the conjunctiva and mouth.14
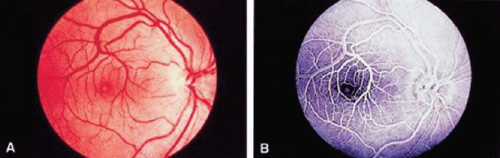 Figure 22-5. A. Superotemporal retinal venous macrovessel traversing the horizontal raphe and a foveal cyst. B. IVFA demonstrates the retinal venous macrovessel and foveal cyst. There is no extravascular leakage. |
The retinal arteries and the accompanying capillary bed are relatively normal in eyes with congenital retinal macrovessels. This is in contrast to congenital retinal arteriovenous communications, which do not feature a normal intervening capillary bed.
ARTERIOVENOUS COMMUNICATIONS
Congenital retinal arteriovenous communications are abnormally dilated retinal vessels that are accompanied by an abnormal or absent capillary system, resulting in a more direct connection between afferent and efferent vessels. They are unilateral and involve either single or multiple sites with a predilection for the papillomacular area and the superotemporal quadrant.36 Both sexes are equally affected. Archer et al37 divided these communications into three types based on size of the affected vessels and condition of the intervening capillary bed. Type I is a typical retinal macrovessel associated with an intervening capillary bed containing abnormally dilated vessels. Archer type II retinal arteriovenous communication consists of a dilated and tortuous artery and vein directly connected, without an intervening capillary bed. Type III arteriovenous communication is an exaggerated version of type II, with the artery and vein so aberrant and intertwined that no distinction between afferent and efferent vessels can be made on ophthalmoscopic examination (Fig. 22-6A). Type II and type III malformations also are referred to as racemose, cirsoid, or arteriovenous aneurysms, although they are not true aneurysms.14,38
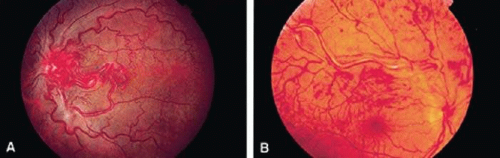 Figure 22-6. A. Archer’s grade III retinal arteriovenous communication (racemose aneurysm) in a patient without the Wyburn-Mason syndrome. Visual acuity was 20/200 (6/60). B. Superotemporal branch retinal vein occlusion associated with a grade III retinal arteriovenous communication (racemose aneurysm). (A and B courtesy of Jerry A. Shields, M.D. and Carol L. Shields, M.D.) |
When arteriovenous communications are large, visual acuity can be severely affected, and the optic nerve tissue can be completely replaced by the anomalous vessels. Vascular sheathing and retinal pigmentary degeneration may occur. Patients with more severe retinal involvement are more likely to have cerebral or periorbital arteriovenous abnormalities, which compose the Wyburn-Mason syndrome.36
The clinical appearance of these lesions may change over time.39,40 Complications associated with this vascular abnormality include intraretinal hemorrhage, vitreous hemorrhage, aneurysm formation, neovascular glaucoma, macular hole, retinal artery occlusion, and branch or central retinal venous occlusion (Fig. 22-6B).41,42,43,44,45 It is hypothesized that retinal arteriovenous communications are associated with localized decreased retinal arterial pressure, increased retinal venous pressure, increased turbulence of blood flow, and decreased perfusion of adjacent retinal tissues to account for these complications.41 IVFA may show rapid dye transit through the arteriovenous communication. There may be adjacent microvascular abnormalities or areas of capillary nonperfusion. Extravascular leakage from these lesions is uncommon. Congenital retinal arteriovenous communications should be distinguished from those acquired secondary to carotid occlusive disease associated with retinal ischemia.46 No successful treatment of these lesions has been reported. Laser photocoagulation may be warranted in some cases with exudative macular involvement or neovascular glaucoma.14,42
A racemose or cirsoid retinal arteriovenous communication may represent the ocular manifestation of Wyburn-Mason syndrome, which is one of the phakomatoses.38 This syndrome consists of unilateral retinal arteriovenous communication in conjunction with an ipsilateral arteriovenous malformation of the midbrain.47 The presence of a concomitant systemic arteriovenous communication correlates directly with the grade of retinal arteriovenous communication. Most grades I and II arteriovenous communications appear to represent isolated retinal vascular anomalies.36 Unlike the other phakomatoses, the hallmark ocular lesion of Wyburn-Mason syndrome is not a true hamartoma but is better classified as a hamartia.38 Additional systemic findings in this syndrome include arteriovenous malformations of the bones of the skull, particularly the maxilla and mandible, and, occasionally, facial angiomas.48 Proptosis secondary to an orbital arteriovenous malformation rarely occurs. Wyburn-Mason47 estimated that 81% of patients with a racemose aneurysm of the retina had an associated intracranial arteriovenous malformation, although other authors believe the actual percentage to be much less.49 No hereditary pattern has been identified.
IDIOPATHIC JUXTAFOVEOLAR RETINAL TELANGIECTASIS
Idiopathic juxtafoveolar retinal telangiectasis (IJRT) is characterized by abnormal retinal capillaries and variable intraretinal exudation after exclusion of an ocular or systemic cause.50,51 Onset is in adulthood, and patients present with blurred central vision. The hallmark lesions are small, perifoveal, and parafoveal telangiectatic capillaries that demonstrate tortuosity, dilation, and irregular caliber and may be associated with lipid exudation in some cases (Fig. 22-7). Microaneurysms can be present.52 The abnormal vessels are either localized to an area that usually is temporal to the fovea and measures between 1 and 5 mm in diameter or scattered diffusely throughout the macula.50,52 Macular edema or lipid exudation can decrease vision.53 An updated classification by Gass and Blodi54 in 1993 divides patients based on biomicroscopic and IVFA features into three groups (1, 2, and 3) and subgroups (A and B). Overall, the telangiectases appear to be related to retinal capillary leakage in group 1, capillary diffusion abnormalities in group 2, and capillary occlusion in group 3.
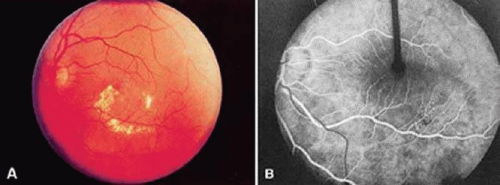 Figure 22-7. A. Fundus photograph of a patient with idiopathic unilateral juxtafoveal retinal telangiectasis. Notice the localized lipid exudate. B. IVFA reveals focal areas of telangiectasis and leakage. |
Group 1A is composed of unilateral congenital parafoveal telangiectasis, typically occurring in young to middle-aged male patients. A small area of easily visible retinal vascular abnormalities is present in the temporal macula. There is mild visual loss from macular edema. Laser photocoagulation to areas of leakage may improve vision. Group 1B consists of middle-aged men with a small area of unilateral idiopathic retinal telangiectasis at the edge of the foveal avascular zone. Laser photocoagulation usually is not performed because of the close proximity to the fovea. Group 1 unilateral juxtafoveal telangiectases may represent a localized form of congenital retinal telangiectasis (Coats syndrome).
In contrast, group 2 consists of bilateral acquired idiopathic parafoveal telangiectases that become clinically apparent later in the fifth and sixth decades.54 The zones of telangiectasis tend to be symmetric and measure up to 1 disc diameter, with a preference for the temporal parafoveal region. Retinal edema and hard exudates are minimal. So-called right angle veins are seen draining the telangiectatic areas. Underlying retinal pigment epithelial alterations and superficial retinal refractile deposits can develop. Some patients develop a small, yellow, pseudovitelliform lesion within their fovea. Visual loss usually is mild but can be severe. Pericentral scotomas are common. Later in the course, choroidal neovascularization (CNV), which often is subfoveal, foveolar atrophy, and intraretinal pigment plaques may develop.51 The initial site of dysfunction in this condition is not known. Histopathologic study of bilateral juxtafoveal telangiectasis reveals endothelial cell and pericyte degeneration, as well as retinal capillary narrowing associated with endothelial cell basement membrane proliferation.55 Gass56 proposed that the primary abnormality may be either the parafoveal retinal neural or Müller cells. In a supporting study, Cohen et al57 examined a large number of patients with group 2a idiopathic juxtafoveal retinal telangiectasis using OCT. They found intraretinal and subretinal pockets of fluid, or cysts, in the fovea, in the absence of foveal thickening. Additionally, there was evidence on OCT of outer retinal atrophy. Other studies of IJRT group 2a using OCT have found similar results.58,59 Cohen et al57 contend that Gass was correct in the proposition that Müller cell dysfunction is what drives the pathology in group 2a IJRT as abnormally functioning Müller cells can account for the spectrum of clinical and OCT findings in this disease.57 OCT may also be useful in the staging of IJRT, though universally accepted staging criteria have not yet been defined.
Severe vision loss in IJRT is most often the result of secondary CNV membrane formation, and until recently, effective therapies for this were not available. Photocoagulation was infrequently used in the past to treat CNV in IJRT because of the proximity of the lesions to the fovea. Surgical removal of subfoveal CNV has had poor results because of its adherence to the overlying neurosensory retina.60 The development of CNV in many different ocular conditions has been found to be related to the production of vascular endothelial growth factor (VEGF), and there has been much success using anti-VEGF medications in the treatment of CNV for various ocular diseases. In the last few years, there have been several published reports of anti-VEGF agents, specifically ranibizumab and bevacizumab, being used to treat CNV in patients with IJRT. Most of the studies show favorable anatomic and functional results, with resolution of the CNV and improvement in the visual acuity.61,62,63,64 The most effective treatment regimens and the long-term success using the anti-VEGF agents, however, have yet to be determined.
Group 3 is an uncommon variant with bilateral prominent idiopathic parafoveal telangiectasis and minimal exudation. Visual loss occurs from progressive parafoveal capillary occlusion. There is no leakage from the capillary bed. These patients may have related systemic disease.
Retinal vascular telangiectasia also can occur in association with some systemic and ocular conditions. Sickle cell disease and bilateral carotid artery obstruction can mimic the idiopathic version of this condition.65,66 Dilated perifoveal capillaries also have been seen with Stargardt disease and facioscapulohumeral muscular dystrophy.67,68 Retinal telangiectasia may occur with retinal vein occlusion or radiation retinopathy or represent an atypical presentation of background diabetic retinopathy.69 In one series, 62% of patients with bilateral juxtafoveolar retinal telangiectasis had abnormal glucose metabolism.70 For this reason, all affected patients should undergo a glucose tolerance test, a fasting blood glucose, or hemoglobin A1c measurement to rule out latent diabetes mellitus.
CONGENITAL RETINAL TELANGIECTASIS (COATS DISEASE)
In 1908, Coats71 published his findings on patients with retinal diseases associated with massive subretinal exudation. He divided the patients into three groups:;i) those with retinal vascular disease, (ii) those without retinal vascular disease, (iii) and those with a large arteriovenous communication. It is accepted that Coats’ third group were patients with retinal capillary hemangiomas (von Hippel angiomas).72 Coats’ first group consisted of patients with the severe form of a disease that Reese73 has since renamed congenital retinal telangiectasis. The term Coats disease should be reserved for patients with congenital retinal telangiectasis associated with marked hard exudation.
Congenital retinal telangiectasis (Coats disease) is an idiopathic retinal vascular disorder that usually affects young male patients unilaterally in their first or second decade of life. Congenital retinal telangiectasis, however, can affect patients of either gender and become manifest at any age. Up to one third of patients are older than 30 years of age at the time of presentation.14 There is no defined familial inheritance. Patients often present with decreased vision, though children may present with strabismus or leukocoria. The hallmark feature of congenital retinal telangiectasis is localized fusiform aneurysmal dilations of the retinal vessels reminiscent of tiny light bulbs73 (Fig. 22-8A). These aneurysms and telangiectatic retinal vessels are often adjacent to areas of retinal capillary nonperfusion with dilated intercapillary spaces. The vascular anomalies can occur anywhere in the fundus and may involve the capillaries, arteries, and veins. Other findings may include vascular loops and beading, retinal neovascularization, hemorrhagic retinal macrocysts, and segmentally dilated capillaries.74 Leakage from the incompetent vasculature may lead to retinal edema, lipid deposition, or, in severe cases, an exudative retinal detachment. The extent of retinal involvement is variable. Infants and children often are more severely affected with extensive vascular involvement and massive subretinal lipid exudate. Vascular leakage may produce a cloudy yellow or green subretinal exudate, which gravitates toward the posterior pole. When the serous component of the exudate resorbs, the yellow exudate remains in the macula and may eventually develop into a fibrovascular macular scar. In contrast, a more localized area of telangiectasis may decompensate in adulthood and present as group 1 idiopathic juxtafoveal retinal telangiectasis. IVFA shows aneurysmal dilation and leakage from telangiectatic retinal vessels adjacent to areas of retinal capillary dilation and nonperfusion (Fig. 22-8B). The clinical course is variable but often progressive. Secondary complications include cataract, vitreous hemorrhage, neovascular or angle-closure glaucoma, CNV, and phthisis bulbi in severe cases. Histopathologic study reveals a characteristic proteinaceous exudate in the subretinal space containing cholesterol clefts and foamy histiocytes. Intraocular calcification rarely has been demonstrated in Coats disease, which is an important consideration when differentiating this entity from retinoblastoma.75
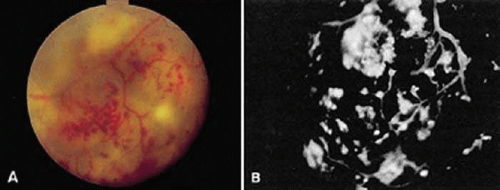 Figure 22-8. A. Unilateral congenital retinal telangiectasis and massive lipid exudation (Coats’ disease) in a 16-year-old boy. Notice the characteristic telangiectatic vessels resembling light bulbs. B. IVFA of the same area revealed hyperfluorescence of the telangiectasia and areas of capillary nonperfusion. |
The syndrome of Leber miliary aneurysms is a form of congenital retinal telangiectasis that is intermediate in severity. Unilateral, congenital retinal telangiectasis, Leber miliary aneurysms, and idiopathic juxtafoveal retinal telangiectasis represent the clinical spectrum of a single primary retinal vascular disorder.76
Treatment of congenital retinal telangiectasis is directed towards closing the leaking telangiectatic vessels to permit resorption of exudate. The treatment modality depends on the location and extent of the lesions.77 Laser photocoagulation is the treatment of choice for small telangiectatic lesions. Cryotherapy may be required for extensive peripheral lesions or for lesions associated with exudative retinal detachments. Multiple treatment sessions may be required. Treatment is directed at the leaking telangiectasis and adjacent zones of capillary nonperfusion. Currently, the VEGF inhibitors are being studied for their possible uses in Coats disease. In the last few years, several published reports have shown successful treatment of Coats with intravitreal bevacizumab, at times in conjunction with intravitreal triamcinolone. Most patients treated with intravitreal bevacizumab demonstrated significant reductions in subretinal fluid and improvement in visual acuity. However, the studies consist mainly of small case series with limited follow-up.78,79,80,81,82 The optimal treatment regimen using the anti-VEGF agents, when they should be used alone or in conjunction with laser photocoagulation, intravitreal triamcinolone or cryotherapy, as well as the long-term success with these new drugs have yet to be determined.
Surgical management with external drainage of subretinal fluid with or without scleral buckling may be necessary if there is a severe exudative retinal detachment.83 Vitrectomy is occasionally indicated for severe or tractional retinal detachments.84 New telangiectasis may appear in areas of retina that were previously uninvolved; therefore, close follow-up is imperative. Despite successful closure of telangiectasis, visual results may be poor, especially when there is macular exudate at presentation.85 Coats syndrome or a Coats-like response (retinal detachment with massive subretinal exudation) has been associated with other ocular and systemic diseases, including retinopathy of prematurity, branch retinal vein obstruction, retinitis pigmentosa, pars planitis, muscular dystrophy, and hemifacial atrophy.54,77,86,87,88,89
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


