Pediatric Ophthalmology
Edited by P. F. Gallin
Thieme Medical Publishers, Inc.
New York ©2000

17

Uveitis
The blood-aqueous and blood–retinal barriers are designed to prevent inflammatory components from entering the eye. When these barriers break down, regardless of the mechanism involved (infectious, autoimmune, etc.), uveitis ensues. Ocular inflammation may occur with or without an accompanying systemic illness. The diagnostic evaluation of uveitis in children therefore involves (1) a careful and detailed ocular and medical history, (2) ocular and systemic examination, and (3) appropriate laboratory tests tailored to the pertinent uveitis history and findings on examination.
 History
History
The relevant history is best understood if one is thoroughly familiar with all the potential diagnostic possibilities.1–4 The reader is encouraged to refer to the specific uveitis syndromes to further understand the relevance of each part of the history taking. The list below outlines all pertinent history that should be obtained.
Obtaining Patient History
Demographics: Establish the age, gender, and ethnicity of the patient. Present illness: Presenting symptoms (if any) such as pain, redness, photophobia, floaters, and decreased vision are established. Determine the onset time and whether the disease is unilateral or bilateral. Of note, although some uveitides may have bilateral involvement, only one eye may be affected during any single inflammatory episode. Preceding events (e.g., trauma, viral illness, or recent immunization) should be determined. What has been the course of the disease including response to anti-inflammatory therapy? Establish the treatment regimens previously used and the degree of patient compliance to determine if previous attempts at treatment were adequate to justify using alternative anti-inflammatory medicines. Ocular history: A history of previous eye disease should be ascertained. Medical history: Is there a history of arthritis or joint pain [juvenile rheumatoid arthritis (JRA), sarcoid, ankylosing spondylitis]? Any history of gastrointestinal symptoms such as abdominal pains, bloody stool, or chronic diarrhea [Inflammatory bowel disease (IBD), Reiters]? Any history of skin disease [Vogt-Koyanagi-Haradas (VKH), psoriatic arthritis, sarcoid, Bechets]? History of exposure to tuberculosis? History of tick bite or hiking in wooded areas (Lyme)? History of any neurological symptoms (VKH, sarcoid, Bechets, multiple sclerosis)? History of urethral discharge (Reiters)? Travel and geographic history: Lyme disease is most common in the mid-Atlantic, New England area. Histoplasmosis is endemic in the Ohio-Missisippi-Missouri Valley region, and Coccidiomycosis-related uveitis is primarily seen in the San Joaquin Valley in California. Cysticercosis, amebiasis, and onchocerciasis are primarily found in Central and South America and Africa. Perhistory: Did patient’s mother have exposure to cats or cat feces during pregnancy (toxoplasmosis)? Did patient have any exposure to dogs or dog feces [e.g., contaminated sandboxes (toxocariasis)]? History of pica? Dietary history: Did patient’s mother ingest undercooked meat during her pregnancy (toxoplasmosis)? Sexual history: Is the patient sexually active? What is the number of sexual contacts? Sexual orientation of the patient? Any penile or vaginal sores? Is there a history of venereal disease? Drug history: Is there a history of maternal intravenous drug use (HIV, fungal infection, CMV retinitis)? Family history: Does mother of patient have a history of active syphilis or HIV infection during pregnancy? Although uveitis is generally not inherited, a family history of uveitis should be sought because certain uveitides have strong HLA associations. |
 Examination
Examination
External examination should be tailored to the elicited history. The joints should be examined for evidence of joint swelling. Look for cutaneous manifestations such as rash, erythema nodosum (subcutaneous erythematous tender nodules most commonly in the anterior tibial region), mucocutaneous lesions, psoriatic lesions, vitiligo, poliosis, alopecia, nail pitting, and onychyolysis. Is seventh nerve function intact (sarcoid or Lyme disease)? The lids and periorbital region should be examined for lacrimal, salivary, or parotid gland swelling and lid margin granulomas. The conjunctiva should be inspected for evidence of sarcoid granulomas, ciliary flush (perilimbal injection), episcleritis, or scleritis. (In scleritis the vascular congestion is localized in the deep episcleral network and does not vasoconstrict with 10% neosynephrine drops.)
Slit Lamp Examination
Anterior Segment Evaluation
Examine the cornea for evidence of dendritic keratitis and check for normal corneal sensation (herpes simplex or herpes zoster-related uveitis). The presence of band keratopathy should be noted. The endothelium should be examined for the presence of keratic precipitates (KPs), which are the most common corneal finding in uveitis patients (Fig. 17-1). The pattern of KP distribution should be noted. KPs usually accumulate in the lower half of the cornea in a base down triangle configuration (Arlt’s triangle). Small KPs with distinct margins are classified as nongranulomatous, whereas larger greasy appearing KPs are classified as granulomatous. The anterior chamber should be inspected for the presence of cells and flare (protein) and the degree of inflammation carefully graded (Table 17-1). Cells and flare can be identified by shining the slit lamp beam obliquely across the anterior chamber while focusing posterior to the cornea (Fig. 17-2). Anterior chamber cells are more easily seen by tipping the light source housing approximately 10 degrees from its normal vertical position. Of note, because chronic flare is generally felt to be a manifestation of chronic damage to the blood-aqueous barrier (and not a sign of active inflammation), flare alone should not be treated. The iris should be assessed for evidence of stromal atrophy and heterochromia. The iris should also be inspected for the presence of nodules. Koeppe nodules are located at the sphincter margin and may be seen in granulomatous and nongranulomatous inflammatory disorders. Bussaca nodules are located on the iris stroma and are generally seen only in granulomatous inflammatory conditions (Fig. 17-3). Because uveitis can produce ischemia, the examiner should look for signs of iris rubeosis (usually evident at the sphincter margin). The presence or absence of posterior synechiae should be noted. A gonioscope or three-mirror lens should be used to look for signs of peripheral anterior synechiae. The angle should also be inspected for the presence of rubeosis, occult foreign body, or malignancy. The lens should be evaluated for the presence of cataract formation. Because glaucoma is a common complication of uveitis and/or the use of steroids the intraocular pressures must always be checked. Alternatively ocular hypotony may be present secondary to chronic inflammation leading to ciliary body shutdown.
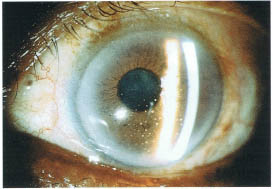
FIGURE 17-1. Slit lamp photograph of granulomatous keratic precipitates in a patient with sarcoidosis.
| No. cells/1 × 1 mm slit beam | Grade |
|---|---|
| 1-2 | Rare |
| 3-4 | lrace |
| 5-10 | 1 + |
| 10-20 | 2 + |
| 20-50 | 3 + |
| Over 50 | 4 + |
a Light intensity and magnification should be maximal.
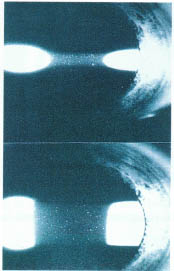
FIGURE 17-2. Slit lamp photograph demonstrating the apprearance of cells and flare.
Vitreous
The vitreous should be inspected for the presence of cells, opacities, snowballs (vitreous cells in clumps), and posterior vitreous separation. Posterior KPs may be seen lining the posterior hyaloid. Cellular activity primarily in the retrolental area (Berger’s space) generally signifies ciliary body involvement. To grade the degree of vitreous inflammation a grading scale based on the qualitative view of the optic nerve and posterior retina compared to standard color photographs is felt to be more useful than trying to grade the number of vitreous cells by retroillumination.5
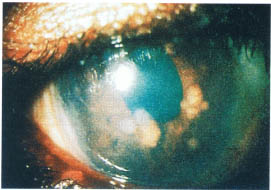
FIGURE 17-3. Slit lamp photograph of Bussaca iris nodules in a patient with sarcoidosis.
Retina and Choroid
The optic nerve should be examined for evidence of hyperemia, papillitis, or papilledema. Of note, granulomas and drusen of the optic nerves can simulate an elevated nerve. The macula is inspected for cystoid macula edema, the most common retinal finding in patients with uveitis (Fig. 17-4). Retinal neovascularization or choroidal neovascularization may occur as a complication of uveitis. The presence of a choroidal neovascular membrane involving the macula is suggested by the finding of subretinal heme lipid or serous detachment. Vascular alterations such as occlusion sheathing, hemorrhages, and cotton wool spots should be noted. Of note, sheathing is more easily seen in the posterior pole compared to the retinal periphery. The retina and choroid are then examined for evidence of focal inflammatory lesions. Exudative nonrhegmatogenous serous retinal detachments may be seen (VKH, scleritis, central serous retinopathy). The retinal periphery should be examined for the presence of a pars plana snowbank, which appears as a white mass usually over the inferior pars plana and adjacent ora serrata region.
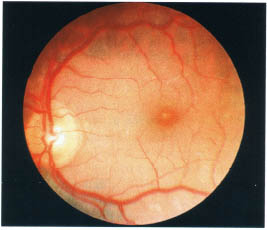
FIGURE 17-4. Fundus photograph demonstrating the appearance of cystoid macula edema.
 Classification of Uveitis
Classification of Uveitis
Several classification schemes may be used to help classify uveitis.
Duration of Inflammatory Activity
Acute uveitis is defined as inflammatory activity that with appropriate intervention lasts less than 3 months, usually between 2 and 6 weeks. The inflammation may recur (acute recurrent), but between episodes the patient is inflammation free.
Chronic inflammation is defined as inflammatory activity that persists beyond 3 months despite appropriate intervention. Periodic exacerbation of the baseline inflammatory activity may occur (chronic-recurrent). Both acute and chronic inflammation may have a sudden or insidious onset.
Severity of Inflammatory Activity
The severity of disease can be graded according to the extent of visual damage. Severe inflammation is defined as greater than 50% visual loss as assessed by visual acuity or electroretinogram, whereas mild inflammation would represent less than a 50% loss.
Anatomic Region of Involvement
Inflammatory activity confined to the anterior chamber is termed iritis. Iritis plus cells in the retrolental space signifying ciliary body involvement is called iridocyclitis. Intermediate uveitis is characterized by inflammation predominantly involving the vitreous and peripheral retina sometimes associated with a peripheral perivasculitis. Posterior uveitis is characterized by inflammation posterior to the vitreous base. Depending on the site of primary involvement, posterior uveitis may be subclassified as focal, multifocal, or diffuse choroiditis, retinitis, or chorioretinitis. When inflammatory findings involve the entire uveal tract the term panuveitis is used.
Response to Steroid Therapy
Inflammatory activity is either responsive or resistant to steroid therapy. If responsive, the maintenance dose needed to control inflammation should be noted.
 Masquerade Syndromes of Anterior and Posterior Uveitis
Masquerade Syndromes of Anterior and Posterior Uveitis
In the pediatric age group masquerade syndromes of anterior and posterior uveitis include rhegmatogenous retinal detachment, intraocular foreign body, and intraocular neoplasm such as retinoblastoma, leukemia, and juvenile xanthogranuloma. Juvenile xanthogranuloma, which may masquerade as anterior uveitis, occurs in children under 15 years of age and is characterized by yellowish-gray iris tumors associated with spontaneous hyphema and anterior segment inflammation. The diagnosis may be established by anterior chamber paracentesis, iris biopsy, and skin examination.
 Noninfectious Causes of Anterior Uveitis
Noninfectious Causes of Anterior Uveitis
Juvenile Rheumatoid Arthritis (JRA)
JRA is a rheumatoid factor negative inflammatory arthritis of at least 3 months’ duration that develops before age 16. Approximately 70% of all childhood arthritis is secondary to JRA. Girls are only slightly more commonly affected than boys, but girls carry a much higher risk of developing uveitis. The peak incidence of JRA occurs between ages 2 and 4, whereas the average age of diagnosis of uveitis is 6 (range 1 to 35 years). If there is no evidence of ocular inflammatory disease within 7 years of the diagnosis of JRA, the subsequent risk of developing uveitis is considered small.6–8
Three subgroups of JRA are identified by the extent of joint involvement that occurs during the first 3 months: systemic onset (or Still’s disease), polyarticular onset, and pauciarticular onset. The risk of developing uveitis is determined by the extent of joint involvement during the first 3 months of the disease and not by the extent of joint involvement at the time of the patient’s first visit.
Systemic onset JRA occurs in approximately 20% of JRA patients. Onset is characterized by fever, lymphadenopathy, hepatosplenomegaly, pericarditis, and a maculopapular rash. Most patients do not develop progressive arthritis. The development of uveitis in this subgroup is so uncommon that annual eye screening for uveitis is adequate.
Polyarticular onset JRA occurs in an estimated 20% of JRA patients. It is characterized by involvement of five or more joints. About one fourth of patients in this subgroup are seropositive for rheumatoid factor. Like adults with rheumatoid arthritis they are not at risk for developing uveitis. The incidence of uveitis ranges between 7 and 14%. Due to the moderate risk of developing uveitis, screening every 6 months is recommended.
Finally, 60% of JRA patients will have pauciarticular onset characterized by involvement of four or fewer joints. The knees and, less frequently, ankles and wrists are most commonly involved. The incidence of uveitis in this subgroup ranges between 78 and 91%. Of note, JRA that is diagnosed as pauciarticular may years later evolve into the polyarticular form. Nevertheless, these patients still carry the risk of developing uveitis associated with the pauciarticular subgroup. Due to the high risk of developing uveitis, eye screening every 3 months is recommended.9 The presence of circulating antinuclear antibodies (ANAs) is especially common in patients with eye involvement. Therefore, in the setting of pauciarticular ANA-positive JRA, routine eye screening is recommended every 2 months (Table 17-2).
Uveitis in JRA is usually asymptomatic, bilateral, and of the chronic anterior nongranulomatous variety. In eyes with severe anterior segment inflammation, cells may also be seen in the anterior vitreous (iridocyclitis). Common complications of uveitis include band keratopathy, posterior synechiae, cataract (incidence of 28 to 31%) glaucoma (incidence of 14 to 22%), and cystoid macula edema.

The treatment of uveitis is designed to eliminate the cellular component of the inflammation and not necessarily the flare that may also be present. Topical steroids (prednisolone acetate 1%) may initially need to be used up to every hour for several days with gradual tapering thereafter. For patients requiring high-frequency dosing of topical steroids or who have not adequately responded to topical steroids, anterior subtenon injections of methylpredisolone and/or a brief course of systemic steroids may be helpful.10 The antimetabolite methotrexate, given weekly in low doses, can be a safe and effective alternative to the use of oral steroids. Other immunosuppressive agents to consider in the treatment of JRA include aza-thioprine, chlorambucil, cyclosporin, and cyclophosphamide. EDTA chelation is used to remove visually significant band keratopathy. To prevent posterior synechiae formation a short-acting mydriatic such as Mydriacyl 1% should be used several times a day. In mild cases one drop at bedtime may be adequate. Cataract surgery should ideally be performed after intraocular inflammation has been controlled for at least 2 to 3 months. In many cases this goal may be difficult to achieve, resulting in a significant delay in surgery, and must be weighed against the possibility of irreversible amblyopia that may develop as a result of the delayed surgery. Lensectomy-vitrectomy through an incision at the limbus or pars plana is the method of choice. Of note implantation of an intraocular lens in JRA patients with chronic uveitis is contraindicated. For control of complicating glaucoma, nonmiotic medical management should initially be employed, although glaucoma filtering surgery with antimetabolites or trabeculodialysis is often eventually required. Macula edema is often secondary to uncontrolled inflammation and is best treated by controlling intraocular inflammation.
Juvenile Ankylosing Spondylitis (JAS)
Childhood juvenile spondylitis is initially characterized by a peripheral lower limb arthritis, which in 15% of cases may be complicated by acute unilateral nongranulomatous anterior uveitis. Early on there may be no sacroiliac involvement, although eventually sacroiliac and lumbosacral spine involvement occurs. The disease typically affects males approximately 10 years of age who carry the HLA B27 tissue haplotype. Rheumatoid factor and antinuclear antibody are generally negative. As in adult ankylosing spondylitis, there is no correlation between disease activity and uveitis.11
Patients are usually symptomatic with blurred vision, pain, and redness. The uveitis is classified as the acute-recurrent type. Although attacks are usually unilateral, both eyes eventually become involved. Signs of acute anterior iritis include nongranulomatous KPs, cells, and flare. Repeated bouts may lead to chronic changes such as band keratopathy, synechiae, and cataract. Treatment involves the use of topical steroids and mydriatics.7
Juvenile Psoriatic Arthritis
Definite JPsA is defined as (1) arthritis associated but not necessarily coincident with a typical psoriatic rash or (2) arthritis plus three of four of the following minor criteria: dactylitis (inflammation of fingers), nail pitting, family history of psoriasis, or a nontypical psoriasis-like rash. Onset of the arthritis is usually pauciarticular with a polyarticular course. Both small and large joints may be involved with prominent knee and digital involvement common. Females are twice as likely as males to be affected. ANA positivity is common but rheumatoid factor (RF) is absent. There may be an association with HLA B-7 antigen. The anterior uveitis that occurs in approximately 15% of children is usually bilateral and chronic. Treatment involves the use of topical steroids and cycloplegics.7,12
Juvenile Reiter’s Syndrome
This syndrome, which is rare in children, includes the triad of arthritis, urethritis, and conjunctivitis.13 An antecedent history of gastrointestinal tract infection caused by gram-negative organisms such as salmonella, shigella, and yersinia is common. It may also follow a nongonococcal urethritis caused by chlamydia or ureaplasma urealyticum. A strong association with HLA B27 haplotype exists. Approximately 2% of patients will develop an acute anterior uveitis. The conjunctivitis is self-limited. Treatment of the anterior uveitis involves topical steroids and cycloplegics.7
Inflammatory Bowel Disease
Inflammatory bowel disease is characterized by a peripheral arthritis in association with regional enteritis (Crohn’s) or ulcerative colitis.14 Ocular findings include episcleritis, scleritis, keratitis, and iritis. The anterior uveitis may be acute or chronic and is more commonly seen with ulcerative colitis (10%) compared to Crohn’s (2%).15 Posterior segment findings may include vitritis, retinal vasculitis, and optic neuritis.
Pediatric Sarcoidosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


