Pediatric Ophthalmology
Edited by P. F. Gallin
Thieme Medical Publishers, Inc.
New York ©2000

22

Retinoblastoma
Retinoblastoma is the most common and most important malignant intraocular tumor of childhood. It is important that the ophthalmologist makes a prompt and accurate diagnosis and refers the patient to an ocular oncologist for appropriate treatment. This chapter considers general aspects, clinical features, pathology, diagnostic approaches, and management of retinoblastoma. A more detailed discussion on these topics can be found in a reference devoted to intraocular tumors.1–4
 General Considerations
General Considerations
Retinoblastoma is the most common intraocular malignancy of childhood and is second to uveal melanoma as the most common primary intraocular malignancy in humans. Although estimates vary, it occurs with a frequency of approximately 1 in 15,000 live births. Approximately 6% of newly diagnosed retinoblastoma cases are familial and 94% are sporadic. All patients with familial retinoblastoma are at risk of passing the predisposition for tumor development to their offspring.1 The details of the genetics and genetic counseling of patients with retinoblastoma are discussed in the literature2 and are beyond the scope of this chapter.
There is no apparent predisposition for race or gender and no predilection for the right or left eye. The tumor occurs bilaterally in 25 to 35% of cases. In the multicentric or bilateral cases the average number of tumors per eye is five with a random distribution between the two eyes. The tumor is diagnosed at an average age of 18 months, with bilateral cases being recognized at an average age of 12 months and unilateral cases at 23 months. In rare instances, the tumor is first recognized at birth, in the teens, or even in adulthood.1,5
 Clinical Features
Clinical Features
Most retinoblastomas present with leukocoria (a white pupillary reflex) (Fig. 22-1). When a retinoblastoma is small, it appears as a flat transparent or slightly white lesion in the sensory retina (Fig. 22-2). Slightly larger tumors are less transparent and appear solid white (Fig. 22-3). As the tumor becomes larger, dilated tortuous retinal arteries and veins supply and drain the tumor (Fig. 22-4). Some retinoblastomas are associated with seeding of tumor cells into the overlying vitreous (Fig. 22-5). Some untreated retinoblastomas show foci of chalk-like calcification that has been likened to cottage cheese (Fig. 22-6). If a tumor is located in the foveal region, the child may develop either exotropia or esotropia. Although most children with strabismus do not have retinoblastoma, it is important that every child with this finding have a careful fundus examination to exclude the possibility of retinoblastoma.
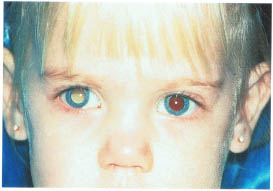
FIGURE 22-1. Leukocoria in a child with retinoblastoma.
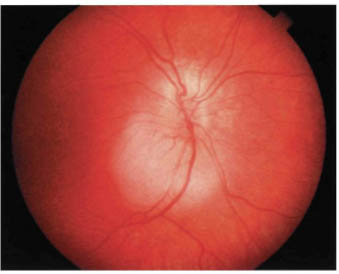
FIGURE 22-2. Small retinoblastoma inferior to optic disc.
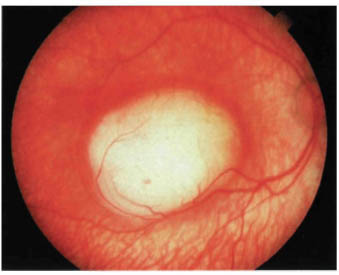
FIGURE 22-3. Slightly larger retinoblastoma showing white color.
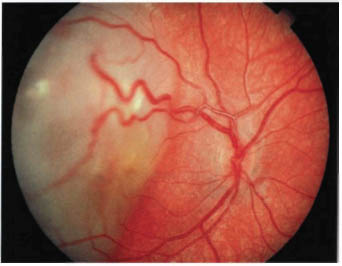
FIGURE 22-4. Retinoblastoma with dilated, tortuous retinal blood vessels.
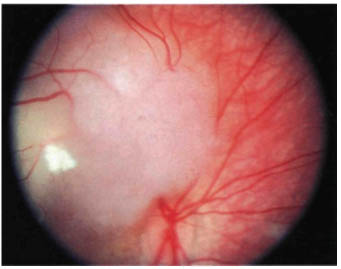
FIGURE 22-5. Retinoblastoma with vitreous seeding.
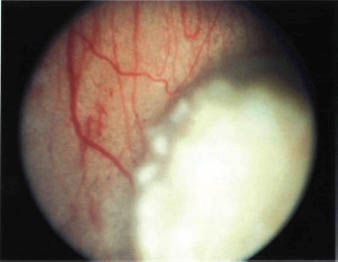
FIGURE 22-6. Retinoblastoma with foci of calcification.
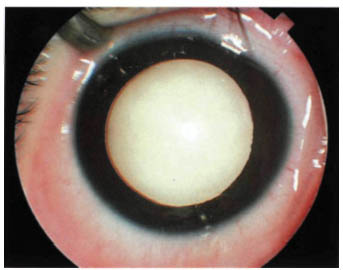
FIGURE 22-7. Endophytic retinoblastoma.
More characteristically, retinoblastoma is larger at the time of presentation and it assumes either an endophytic or an exophytic growth pattern. Such larger tumors almost always cause leukocoria. An endophytic retinoblastoma is one that grows from the retina in toward the vitreous cavity. Hence, it is characterized by a white hazy mass over which no retinal vessels can be visualized (Fig. 22-7). Because of their friable nature, endophytic tumors can eventually seed the entire vitreous cavity and simulate endophthalmitis. An endophytic retinoblastoma can also seed into the anterior chamber and produce multiple nodules at the pupillary margin. With time, the cells may settle into the inferior portion of the angle and resemble a hypopyon.
An exophytic retinoblastoma is one that grows from the retina out into the subretinal space. In contrast to an endophytic tumor, the retinal vessels are apparent on the surface of the tumor. Such tumors produce a progressive retinal detachment, with the retina often displaced anteriorly behind the clear lens and a white mass immediately behind the detached retina (Fig. 22-8). An exophytic retinoblastoma can clinically resemble Coats’ disease or other forms of exudative retinal detachment.
Diffuse infiltrating retinoblastoma is a less common form of retinoblastoma characterized by a relatively flat infiltration of the retina by tumor cells.6,7 Because an obvious mass is not present, there is often a delay in diagnosis. Therefore, diffuse retinoblastomas are usually recognized clinically at an older age than are typical cases of retinoblastoma. They frequently produce vitreous and anterior chamber seeding, which may be confused with intraocular inflammation. Fortunately, almost all reported cases of diffuse infiltrating retinoblastoma have been unilateral sporadic cases with a negative family history. Because of the extensive intraocular seeding in most instances, enucleation has been considered to be the best management.
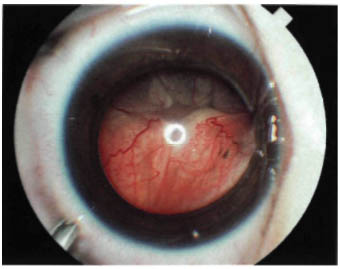
FIGURE 22-8. Exophytic retinoblastoma.
Iris neovascularization (rubeosis iridis) occurs in 17% of all children with retinoblastoma and in about 50% of eyes with advanced retinoblastoma that require enucleation.8 We believe that iris neovascularization usually accounts for the acquired heterochromia iridis that characterizes some cases of retinoblastoma. Any infant with unexplained acquired heterochromia should be evaluated for possible retinoblastoma. Spontaneous bleeding from these vessels may cause a hyphema.
Some necrotic retinoblastomas can produce severe secondary periocular inflammation, resulting in a clinical appearance of preseptal cellulitis or endophthalmitis.9,10 In such cases computed tomography (CT) can reveal a large calcified intraocular mass with periocular soft tissue density suggesting extraocular extension of retinoblastoma. However, these advanced cases usually do not have evidence of extraocular extension after enucleation of the affected eye. The periocular inflammation seen with retinoblastoma appears to be secondary to necrosis within the tumor and not secondary to extraocular extension of the tumor. Tumors with true orbital extension are more likely to show extension to the brain and metastasis to regional lymph nodes.
Trilateral Retinoblastoma
In recent years it has been recognized that some children with the familial form of retinoblastoma can also develop a pinealoblastoma.11,12 Because the pineal tumor has many similarities to retinoblastoma from embryological, pathologic, and immunologic standpoints, the pineal tumor can be detected with CT or magnetic resonance imaging (MRI). The prognosis for life is guarded. Most children who die from retinoblastoma have some degree of intracranial involvement, usually secondary to direct spread through the optic nerve or subarachnoid space. We believe that some earlier reported cases of presumed brain metastasis probably represented pinealoblastoma (trilateral retinoblastoma) that were misdiagnosed as metastatic retinoblastoma before the entity of trilateral retinoblastoma was recognized.
Retinocytoma
Recent evidence supports the existence of an uncommon benign variant of retinoblastoma that has been termed retinoma13 or retinocytoma.14 We believe that the term retinoma is too general because it could be interpreted to mean any tumor of the retina. Although no terminology is perfect, there is a stronger argument forusing either the term retinocytoma or spontaneously arrested retinoblastoma to define this condition.1 A retinocytoma carries the same genetic implications as an active retinoblastoma.13,14
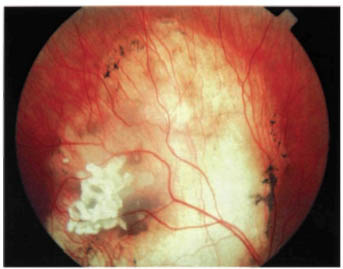
FIGURE 22-9. Spontaneously regressed retinoblastoma in an eye with good vision.
Spontaneously Regressed Retinoblastoma
Complete spontaneous necrosis leading to regression and a “cure” is a well-known phenomenon that is said to occur more frequently in retinoblastoma than in any other malignant neoplasm.1 It is characterized by a severe inflammatory reaction in an eye with retinoblastoma, sometimes followed by the development of phthisis bulbi. In cases of spontaneous regression of a smaller retinoblastoma, the eye may retain good vision (Fig. 22-9). It is not certain whether such tumor regression occurs secondary to vascular ischemia to the tumor or whether more complex immunopathologic mechanisms play a role. In any child with a phthisical eye of uncertain cause, the diagnosis of spontaneously regressed retinoblastoma should be considered. Spontaneously regressed retinoblastoma carries the same genetic implications as an active retinoblastoma.
 Pathology
Pathology
In most cases, retinoblastoma can be readily recognized in the sectioned eye by its typical appearance (Fig. 22-10
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


