Pediatric Ophthalmology
Edited by P. F. Gallin
Thieme Medical Publishers, Inc.
New York ©2000

10

Neuro-Ophthalmology
In evaluating a child with neuro-ophthalmic symptoms and signs, it is useful to divide the involved neural systems into afferent and efferent pathways. However, it should be recognized that many disease processes do not limit themselves to one of these single pathways. In general, disorders of the afferent visual system in children present as visual loss or as the motor consequences of visual loss (i.e., nystagmus in bilateral congenital cataracts). Efferent disorders, on the other hand, present as misalignment of the ocular axis or as impairment of ocular movement. In young children efferent disorders may present without the obvious complaints of diplopia or oscillopsia as these symptoms are conspicuously absent until the age of 5 or 6 years. That children with neurological disorders rarely present with localizing symptoms makes the neuro-ophthalmic examination critically important in arriving at the correct diagnosis.
 Pediatric Neuro-ophthalmic Examination
Pediatric Neuro-ophthalmic Examination
Because children usually cannot describe ocular symptomatology, accurate diagnosis depends on a thorough history obtained from the parent or caretaker and a neuro-ophthalmic examination tailored for children. The history of the illness should focus on onset, duration, variability, frequency, and progression of the ocular sign, whether it is decreased vision or abnormal eye movement. Associated symptoms such as headache, nausea, vomiting, sleep problems, polyuria, polydypsia, and clumsiness should be elicited. Medical history is vital, with obstetrical and birth history often providing clues to the diagnosis (i.e., congenital Horner’s syndrome associated with brachial plexus injury). It must be emphasized to the parents that, although this information is important, most congenital anomalies cannot be attributed to the actions of the parents, and guilt is not being ascribed. The parents must be questioned about the child’s physical, motor, and intellectual development. Delayed development or loss of motor milestones points to generalized neurological disorder. The examiner must be familiar with the norms for attainment of these milestones. A history of preceding or coexistent trauma or generalized illness should be elicited if present. Chronic medication use must be determined because medications can sometimes result in neuro-ophthalmic conditions (i.e., Dilantin toxicity and ethambutol neuropathy). The social and family history cannot be forgotten as there are autosomal dominant forms of optic atrophy, nystagmus, and neuropathy. The examiner must always consider psychogenic factors that may present as sudden loss of vision in children with no objective findings.
The neuro-ophthalmic examination of a child begins the moment the child enters the examination room. Is the child visually attentive to the new surroundings? Does the child interact with the caretaker appropriately and show motor skills that are age appropriate? Is the child’s body or head position abnormal? Once these observations are made, formal examination can begin. The topical diagnosis of a neuro-ophthalmic disorder is made by knowing the visual acuity, visual fields, pupillary behavior, ocular movement, and optic nerve appearance.
Assessment of visual acuity must be conducted by using techniques that are age appropriate. In infants, visual fixation behavior can be assessed binocularly and monocularly. An infant with poor vision will object to occlusion of the sound eye. Acuity can be objectively recorded using visually evoked potential (VEP) or forced preferential-looking (FPL) tests, but both techniques require specialized equipment and well-trained interpreters. Verbal preliterate children can be tested using HOTV, Allen cards, Lea cards, or tumbling E. Older children should be tested with full-line Snellen acuity optotypes. Although subtle visual field changes cannot be quantitated in young children, hemianopias can readily be detected by a two-examiner technique. One examiner sits in front of the child, who is seated on the parent’s lap. Fixation is maintained using an interesting toy. The second tester stands behind the child and introduces a brightly colored toy into the child’s peripheral field. A child with normal peripheral vision will turn toward the second object, and a child with a hemianopia will ignore the object.
Examination of the pupils can be difficult in children because they will not often maintain fixation for prolonged periods of time. Anisocoria is easy to determine in light-colored eyes, and pupil size should be noted in both dim and full light. Fixation on the toy at distance should be assured when checking pupillary reaction to light and when looking for afferent pupillary defect. If fixation is not maintained, pupillary constriction associated with convergence may confound reaction to light. A normal pupillary response provides information about both efferent and afferent systems.
Assessment of the oculomotor system should include an evaluation for strabismus, ductions and versions, and abnormal ocular movements. When examining a child for strabismus, fixation must be maintained using a visually interesting accommodative target. The cover-uncover test is used to detect tropias, and the prism-cover test is used to quantitate the degree of deviation. In younger children, the Hirschberg corneal light reflex test should be used to detect strabismus. Ductions are evaluated by having the child follow a moving target. If a child is not visually attentive, it is still possible to test the efferent visual pathways by utilizing the vestibular ocular reflex. This can be performed by holding the infant facing the examiner and then rotating the child at arm’s length. This will result in conjugate slow deviation with a quick phase refixation. By rotating the child in both directions, cranial nerves III and VI can be evaluated. This is particularly useful in infants with congenital esotropia with abduction deficit.
Although many neuro-ophthalmic conditions will be readily diagnosed and treated by the ophthalmologist, neurological disorders are often best treated using a multidisciplinary approach. A consultation with a pediatric neurologist, neuroradiologist, endocrinologist, neurosurgeon, oncologist, and developmental pediatrician should be obtained when indicated.
Approaching the Blind Infant
Obtaining an objective visual acuity measurement is difficult in children less than 2 years of age. Poor vision in this group can be caused by lesions anywhere in the visual pathway. However, depending on the etiology, visual prognosis can range from normal (delay of visual maturation) to complete blindness. Parents are the most important source of information regarding the visual function of their infant. Questions such as Can your baby see and respond to (smile or follow) your face? and Does your infant respond when you turn on or off the room lights? should be asked. Infants normally fixate on a face by eight weeks of age.
Visual function in infants can be measured in various ways. Because most of these techniques involve interpretation of motor responses, i.e., FPL, fix and follow “visual behavior,” central steady maintained (CSM), the observer must consider and exclude abnormalities in the efferent motor pathways. Despite the dependence on motor pathways, assessing visual function in infants and toddlers primarily relies on the observation of behavior. Guidelines have been established to assess normal visual development. Preverbal children who fail to meet these visual milestones or present with nystagmoid ocular movements should undergo a complete ophthalmic and neurological examination.
| Milestones of Visual Behavior |
|---|
| Steady fixation on [mother’s] face: days to 6 weeks |
| Fixation and following and reaction (smile)to mother’s face: 2 months |
| Visually directed reaching: 4 months |
| Pointing to desired objects: 12 months |
Optokinetic nystagmus (OKN) testing, which is also dependent on efferent pathways, may be helpful in determining an infant’s visual function. To elicit a jerk nystagmus (which is the normal response to an OKN stimulus), the infant must have visual function. Normal vertical OKN responses in an infant with horizontal nystagmus is said to be predictive of visual function sufficient to attend regular schools.
Psychophysical tests such as FPL and VEP have been employed to quantify visual responses in preverbal children. Although these techniques have been useful in studying the visual development of normal infants, quantification of functional vision in the visual and neurologically handicapped infants has proved to be highly variable and should not be employed to predict vision.
Evaluation of the visually impaired infant begins with the general observation of the infant’s tone, head control, and signs of dysmorphism. By 6 weeks of age, an infant should be able to lift the head when prone and smile. At 3 months, the infant should be able to hold objects with the hand and coo. At 6 to 8 months, an infant should be able to sit, roll over, and feed itself. By 12 months of age, the infant should be able to crawl, stand with an aid, and begin to walk.
A complete ophthalmic examination including cycloplegic refraction and ophthalmoscopy is then performed. The ophthalmic exam is geared toward (1) identifying clarity of the visual axis (i.e., excluding anterior dysgenesis, cataract, persistent hyperplastic primary vitreous [PHPV], etc.), (2) assessing the status of the retina (i.e., degree of retinal pigment epithelium [RPE] pigmentation, foveal reflexes, colobomas, falciform folds, etc.), and (3) evaluating the size, morphology, and color of optic nerve (optic nerve hypoplasia, atrophy, or malformation).
Retinoscopy is not only critical to evaluate refractive status but is very sensitive in identifying media (clarity) problems. In the context of a normal ophthalmic examination, a significant refractive error may indicate a syndrome or retinal dystrophy.
| Associations with Refractive Status |
|---|
| Myopia |
| Prematurity and retinopathy of prematurity |
| Vitreoretinal degeneration (Stickler’s syndrome and Wagner’s dystrophy) |
| Retinal syndromes (Bassen-Kornsweig syndrome and Laurence-Moon-Bardet-Biedl syndrome) |
| Albinism |
| Collagen disorders (Ehlers-Danlos syndrome and Marfan syndrome) |
| Other syndromes (Alport’s syndrome, Alagille syndrome, Down syndrome, Fabray’s disease, Flynn-Aird syndrome, and Marshall’s syndrome) |
| Hyperopia |
| Albinism |
| Microphthalmic syndromes |
| Leber’s congenital amaurosis |
| Retinitis pigmentosa (dominant) |
Nystagmus in the visually impaired infant is an important clue to distinguishing anterior from posterior visual pathway pathology. Nystagmus manifests between 2 and 3 months of age in the congenitally blind infant or 1 month following acquired visual loss in an infant less than 2 years of age (nystagmus is not present at birth nor is it acquired as a consequence of visual loss after 2 years of age). Nystagmus in this age group (less than 2 years) almost always indicates a defect in the sensory visual system (even in the infant with a normal ophthalmic examination). Roving eye movement, a type of nystagmus, is characterized by slow, large amplitude “to and fro” eye movements, which are indicative of very poor visual function and prognosis.
As stated earlier, the presence of nystagmus in an apparently blind infant points to an afferent defect in the visual system. Opacities and malformations of ocular structures will be manifest. If optic atrophy or abnormal disc configuration is detected, neuroimaging should be performed. If the exam is normal, an electroretinogram (ERG) should be obtained to rule out Leber’s congenital amaurosis or cone/rod dystrophy.
When nystagmus is absent, the blindness may be cortically mediated due to cortical visual impairment (CVI) or delayed visual maturation (DVM). Neuroimaging is useful in both of these conditions. It is important not to confuse congenital ocular motor apraxia with blindness because these patients cannot generate saccades voluntarily but do have excellent visual acuity despite the appearance of visual inattention.
 Disorders of the Afferent Visual Pathway System
Disorders of the Afferent Visual Pathway System
Congenital Optic Disc Anomalies
Many infants presenting with low vision and nystagmus will have bilateral optic disc abnormalities. Unilateral cases typically present later in life with strabismus (esotropia) and without nystagmus. Disc anomalies are important to recognize due to their association with central nervous system abnormalities. Visual acuity is highly variable and is typically much better than predicted based on optic disc appearance. Structural anomalies may coexist with amblyopia, and every attempt should be made to correct associated refractive errors and patch accordingly.1
| Congenital Optic Disc Anomalies |
|---|
| Optic nerve hypoplasia |
| Morning glory disc |
| Optic disc coloboma |
| Peripapillary staphyloma |
| Optic disc pits |
| Optic nerve head drusen |
| Congenital tilted disc syndrome |
| Optic disc pigmentation |
| Myelinated nerve fiber layer |
Optic Nerve Hypoplasia
Hypoplasia of the optic nerve is a developmental anomaly characterized by a diminished number of axons in the context of a structurally normal optic nerve (Fig. 10-1). Vision is highly variable and is dependent on the number of intact neurons. Visual acuity ranges from 20/20 to no light perception. There may also be generalized or segmental visual field loss.
Optic nerve hypoplasia is caused by a gestational central nervous system injury that results in transsynaptic degeneration and neuronal migration defects. The most common known etiologies are the maternal ingestion of drugs (neuroleptics, alcohol, cocaine, and LSD) during the first trimester and maternal diabetes, but in most cases no known cause can be identified. Optic nerve hypoplasia is also associated with young maternal age.2
Optic nerve hypoplasia is associated with a variety of midline central nervous system defects. In septooptic dysplasia (de Morsier syndrome), there is an associated thinning or absence of the septum pellucidum and corpus callosum. Forty-five percent of patients have cerebral hemispheric abnormalities caused by injury or migration defects (holoprosencephaly, schizencephaly, or periventricular leukomalacia), whereas 15% have posterior pituitary ectopia caused by maldevelopment of the pituitary infundibulum. Therefore, endocrine function must be monitored carefully. Neuroimaging should be performed on children who fall off the growth curve (growth hormone deficiency) or have a history of neonatal jaundice or neonatal hypoglycemia. Neonatal jaundice is associated with congenital hypothyroidism, whereas neonatal hypoglycemia is associated with generalized pituitary dysfunction.3 Due to a suppressed pituitary-adrenal axis, these children are at risk for sudden death during febrile illnesses.4
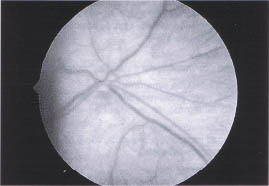
FIGURE 10-1. Optic nerve hypoplasia. Note small amount of nerve tissue surrounded by pigmented epithelium (double-ring sign).
Morning Glory Disc
The morning glory anomaly is usually unilateral and is characterized as a large, funnel-shaped optic nerve with multiple peripheral radial retinal vessels and a central glial tuft. Peripapillary RPE mottling is common and is related to associated serous retinal detachments, which may spontaneously resolve. Like other disk anomalies, visual acuity varies from 20/20 to no light perception, but low vision is the rule. Transspheniodal encephalocele should be suspected in children with morning glory and midfacial anomalies such as hypertelorism and clefting syndromes.5
Optic Disc Coloboma
Colobomas of the optic disc represent a defective closure of the fetal fissure and usually affect the inferior part of the retina and choroid. Mild forms are limited to an inferior excavation of an otherwise normal optic nerve. More profound defects extend into the inferior choroid (including the ciliary body and iris) and retina. In the extreme case, microphthalmos with or without cyst may result. Serous retinal detachments are common with high spontaneous reattachment rates. Colobomas are bilateral in 50% of cases and may be inherited in an autosomal dominant or sporadic fashion.1 Unlike the morning glory anomaly, optic disc colobomas are associated with multisystem genetic disorders.
| Systemic Conditions Associated with Optic Disc Colobomas |
|---|
| CHARGE association (coloboma, heart defects, atresia choane, mental retardation, genito-urinary abnormalities, ear defects/deafness) |
| Goltz syndrome (X-linked dominant, focal dermal hypoplasia) |
| Lenz microphthalmia syndrome (X-linked, microphthalmos with or without coloboma, mild mental retardation, large ears) |
| Meckel-Gruber syndrome (autosomal recessive, coloboma, renal abnormalities, occipital encephalocoele) |
| Walker-Warburg syndrome (autosomal recessive, hydrocephalus, encephalocoele, retinal dysplasia) |
| Goldenhar syndrome (lid coloboma, epibulbar dermoid, ear abnormalities) |
Peripapillary Staphyloma
This very rare unilateral peripapillary excavation is associated with RPE changes. Despite a relatively normal appearing optic nerve, visual acuity is invariably depressed due to cecocentral scotomas.
Optic Disc Pits
Optic pits are unilateral ovoid depressions of the optic disc that are typically located on the temporal margin. Over half the pits have an associated cilioretinal artery arising at their base. Subretinal fluid accompanied by decreased vision (optic pit maculopathy) is common (50%). Spontaneous reattachment occurs in 25% of cases, and the remaining cases may benefit from vitrectomy followed by internal gas tamponade. Due to the schisis nature of the retinal defect, laser photocoagulation does not appear to improve the condition. In the absence of subretinal fluid, visual acuity is not affected. Visual field findings include an enlarged blind spot and arcuate scotomas. Optic pits may represent a mild optic disc coloboma; however, their temporal location and lack of associated genetic syndromes cast doubt on this relationship.
Optic Nerve Head Drusen
Optic nerve head drusen are relatively common (0.3%) and can be confused with papillaedema (Fig. 10-2.). They are bilateral (86%) and associated with cupless optic nerves, visual field defects, transient visual loss, peripapillary sub-retinal neovascular membranes (which are usually self-limited), and peripapillary retinal and subretinal hemorrhage. Optic nerve head drusen can be differentiated from papilledema because (1) disc elevation is confined to disc, (2) there is a lack of venous congestion, (3) the peripapillary nerve fiber layer is not swollen and opaque (i.e., the peripapillary retinal vasculature is readily seen), and (4) ultrasound images show characteristic reflective images.1
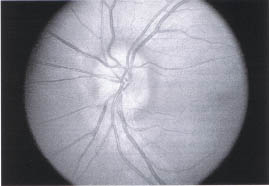
FIGURE 10-2. Optic nerve head drusen. Drusen in optic nerve cause elevation of disc but no edema or obscuration of capillaries.
Congenital Tilted Disc Syndrome
The congenital tilted disc is characterized as an oval disc with an elevated superonasal rim and depressed inferotemporal rim. The tilted disc is associated with situs inversus of the retinal vessels, high myopia with astigmatism, and bitemporal visual field defects that do not respect the vertical meridian.
Optic Disc Pigmentation
A dark disc may represent melanin deposition on the lamina cribosa characteristic of Aicardi’s syndrome, RPE hyperplasia following papillitis, or neoplasm as seen in melanocytoma and RPE hamartomas. Aicardi’s syndrome is an X-linked lethal condition (seen only in heterozygous girls) characterized by infantile spasms, severe mental retardation, agenesis of the corpus callosum, and typical fundus findings. The disc is often pigmented and dysmorphic with multiple peripapillary lacunae.6
Myelinated Nerve Fiber Layer
Myelination of the nerve fiber layer anterior to the lamina cribosa occurs in approximately 1 % of the population. It is bilateral in 20% of cases. The white fluffy fibers usually radiate from the disc but discontinuous patches are not uncommon (Fig. 10-3). Extensive diffuse unilateral myelination has been associated with high myopia and amblyopia that is often refractive to occlusion therapy.1
Optic Atrophy
Optic nerve atrophy is most commonly caused by an injury to the anterior visual pathway with subsequent retinal ganglion cell and axon loss. Prior to birth, posterior visual pathway lesions have been implicated though the process of transsynaptic degeneration. The atrophic optic nerve is diffusely or segmentally pale with associated retinal nerve fiber layer loss. Approximately half the cases of optic atrophy are caused by tumors (29%) and inflammation-related insults (meningitis and optic neuritis) (17%). The remaining etiologies include trauma (11%), idiopathic causes (11%), hereditary causes (9%), perinatal problems (9%), hydrocephalus (6%), and neurodegenerative causes (5%). Rarely (1%), toxic or metabolic etiologies are identified. Hydrocephalus or anterior visual pathway tumors should be suspected in an otherwise normal child with optic atrophy and negative perinatal, medical, and family histories, whereas intracranial, genetic, and neurometabolic etiologies are common in children with associated neurological findings. Optic atrophy is a leading cause of low vision in these mentally handicapped children. Unilateral, asymmetric, and band optic atrophies are highly suggestive of compressive lesions.7
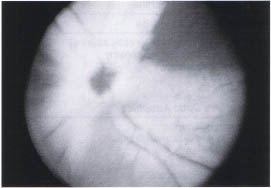
FIGURE 10-3. Myelinated nerve fiber layer. Myelin obscures detail of optic nerve head and retinal vasculature.
Based on etiology, optic atrophy can be divided into three categories: (1) hereditary and neurodegenerative, (2) compressive, and (3) noncompressive. Except for dominant optic atrophy, hereditary and neurodegenerative optic atrophies typically present to the ophthalmologist as part of a multisystem work-up. Children with compressive and noncompressive forms of optic atrophy may present directly to the ophthalmologist as a consequence of abnormal eye movements (nystagmus or strabismus) or failed vision screening exam.
| Causes of Optic Atrophies |
|---|
| Hereditary Optic Atrophies |
| Dominant optic atrophy (Kjer optic atrophy) |
| Leber’s hereditary optic neuropathy |
| Recessive optic atrophy |
| Behr optic atrophy |
| DIDMOAD |
| Compressive Optic Atrophies |
| Craniopharyngioma |
| Optic nerve and chiasmal gliomas |
| Pituitary adenoma |
| Meningioma |
| Sphenoid sinus mucocele |
| Osteopetrosis |
| Craniosystonosis |
| Fibrous dysplasia |
| Noncompressive Optic Atrophies |
| Hydrocephalus |
| Perinatal hypoxia/ischemia |
| Toxic/nutritional |
| Retinal degenerative |
Hereditary and Neurodegenerative Optic Atrophies
Dominant Optic Atrophy
Dominant optic atrophy, or Kjer optic atrophy, has been localized to chromosome 3 and is the most common inherited optic atrophy with an incidence of 1:50,000 births. These children usually present between the ages of 4 and 8 years subsequent to a failed visual screening exam. Visual loss is insidious over the first decade with subsequent gradual deterioration. Children do not have nystagmus and typically have better than 20/80 visual acuity associated with blue-yellow dichromatopsias. Approximately 10% of affected children also have sensoral neural hearing deficits. By age 60 years, visual acuities are typically in the 20/800 range.8,9
Leber’s Hereditary Optic Neuropathy
Patients with Leber’s hereditary optic neuropathy (LHON) usually present in their second to third decades with asynchronous bilateral central loss of vision to the 20/200 level. LHON is a mitochondrial (maternally) inherited disease that primarily affects males. Prior to the acute phase, the retinal examination may reveal pseudoedema of the disc with surrounding telangiectatic microangiopathy. Several point mutations have been implicated, and genetic testing is available at several academic institutions.
Recessive Optic Atrophy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


