Tumors of the Uveal Tract
Carol L. Shields
Jerry A. Shields
The uvea comprises a luxurious vascular bed plus melanocytes, long and short ciliary nerves, and occasional lymphocytes. Each of these components can contribute to the development of a tumor. In addition, the uvea is occasionally involved with metastatic, leukemic, and lymphomatous infiltration due to its rich vascular tissue and high flow rate.
It should be realized that the most common tumor of the uvea is the melanocytic nevus. It has been estimated that uveal nevi affect approximately 5% to 10% of the Caucasian population.1,2,3 On the other hand, the most common malignancy of the uvea is choroidal metastasis. It has been estimated that approximately 2% to 9% of patients with known cancer have or develop uveal metastases.4,5 Given the knowledge that there are approximately 1,400,000 new cancers diagnosed in the United States each year, it is estimated that approximately 26,000 uveal metastases occur, but most of these are not diagnosed due to overriding systemic metastases and life-threatening problems. The most common primary intraocular malignancy is uveal melanoma, of which there are approximately 2,500 new cases per year in the United States. Other tumors such as choroidal hemangioma, choroidal osteoma, and pigment epithelial tumors are much rarer.6
MELANOCYTIC TUMORS OF THE ANTERIOR UVEA (IRIS)
Tumors of the iris can arise from the iris melanocytes, iris smooth muscle cells, and iris pigment epithelium. Systemic neoplasms such as lymphoma and metastatic tumors can also affect the iris. Melanocytic nevus, malignant melanoma and pigment epithelial cysts, and solid tumors will be considered here.
Nevus
An iris nevus is a benign tumor that arises from the melanocytes in the iris stroma.7 In a review of 1,519 iris tumors, nevi represented 723 (69%) of all iris lesions8 (Table 1). Although most iris nevi remain clinically stationary, they can occasionally give rise to malignant melanoma.9 Histopathologically, an iris nevus is usually composed of low-grade cells with spindle configuration. Occasionally an iris nevus is classified as a melanocytoma, when it is composed of deeply pigmented, plump cells with large amounts of cytoplasm and small nuclei that lack mitosis.
TABLE 1. Classification of 1519 Consecutive Iris Tumors Referred for Evaluation on the Ocular Oncology Service at Wills Eye Hospital, Philadelphia | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
An iris nevus classically appears as a variably pigmented, well-circumscribed lesion within the iris stroma (Fig. 1). It can involve any portion of the iris from the pupillary border to the iris root and can be flat or minimally elevated. Occasionally, an iris nevus occupies an entire sector of the iris from the pupillary border to the anterior chamber angle. In such cases, it is suspected to be congenital and even could represent a form of sector melanocytosis. In some cases, iris nevus is clinically amelanotic, and intrinsic corkscrew vessels are apparent within the lesion. An iris nevus can induce an irregular pupil, (corectopia) with the pupillary margin dragged toward the nevus, or it can induce ectropion iridis due to slow, chronic dragging from contractile myofibroblasts. In some cases, sector cortical cataract, slight dusting of pigment in the anterior chamber angle, and minimally dilated episcleral vessels can be found.
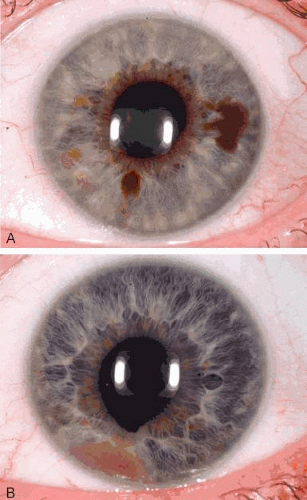 Fig. 1. Spectrum of iris nevus. A: Pigmented iris nevi. B: Nonpigmented iris nevus with corectopia and ectropion iridis. |
Iris nevus is best diagnosed by recognition of the typical lesion with slit lamp biomicroscopy. Ancillary diagnostic studies such as fluorescein angiography can delineate the feeder and intrinsic vascularity. Ultrasound biomicroscopy (UBM) can establish the posterior border and internal echogenic qualities. In recent years, transillumination of the globe and UBM have assumed an important role in defining the posterior extent of iris nevi with angle involvement. In some instances ciliary body extension is found. In other instances, intratumoral cavities on UBM have been noted. Anterior segment optical coherence tomography can also provide information on the internal qualities of nevi but shows less detail about the ciliary body relationship.
The management of iris nevus includes documentation of the mass with accurate drawings and photographs and examination on a 6 month or yearly basis, looking for evidence of related glaucoma or tumor enlargement. Iris melanocytoma has a tendency to undergo necrosis and produce pigmentary glaucoma. If growth of an iris nevus is detected, then malignant transformation into melanoma should be suspected. In an analysis of 175 patients with suspicious iris nevus, growth into melanoma was detected in 5% by 5 years.9 Factors related to growth included superior location on the iris and tumor seeding onto the iris stroma or into the anterior chamber angle.9
Melanoma
An iris melanoma is a malignant tumor that arises from the melanocytes of the iris stroma.7 Histopathologically it is composed of spindle cells, epithelioid cells, or a combination of the two (mixed cell type). Of the approximately 2,500 uveal melanomas each year in the United States, only 2% originate in the iris, and the other 98% originate in the ciliary body or choroid.
An iris melanoma is characterized clinically as a variable pigmented, elevated, circumscribed or diffuse melanocytic neoplasm that affects the iris stroma.10 It is typically larger than an iris nevus and with more dilated feeder and intrinsic vessels, as well as more profuse tumor seeding. The lesion most often is deeply pigmented and elevated (Fig. 2). In can be amelanotic, or it can be partially pigmented and partially nonpigmented.
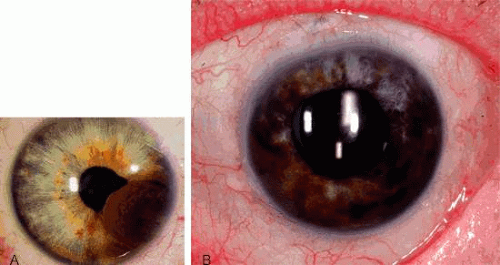 Fig. 2. Iris melanoma. A: Nodular iris melanoma. B: Diffuse iris melanoma. |
An important variant is the diffuse iris melanoma.11 It grows as a flat, diffuse, often multifocal mass that covers a large area of the iris surface. The patient with a diffuse iris melanoma typically presents with a clinical syndrome of progressive acquired hyperchromic heterochromia and ipsilateral secondary glaucoma.11,12
The diagnosis of iris melanoma is best made by recognition of the typical slit lamp features by an experienced observer. Transillumination of the globe, UBM, optical coherence tomography, and fluorescein angiography can add useful information regarding tumor extent, ciliary body extension, tumor vascularity, and internal tumor qualities. Fine-needle aspiration biopsy can be employed to confirm the diagnosis of diffuse iris melanoma in cases where plaque radiotherapy or enucleation is being considered.13,14,15,16
Once the diagnosis of iris melanoma is clearly established, usually by documentation of progressive growth, tumor management is considered. Lesions confined to the iris and with minimal or no seeding can be managed by tumor excision using no-touch technique and partial sector or peripheral iridectomy for those limited to the iris, iridocyclectomy for those with cililary body extension, or iridogoniocyclectomy for those with ciliary body and angle involvement. Complete tumor excision is critical. Plaque radiotherapy is applied if there is evidence of residual tumor or high-grade tumor cell type on pathology.15,16 The diffuse iris melanoma is often too large to resect locally and may require enucleation or plaque radiotherapy in selected cases.5,15,16
Iris Pigment Epithelium Cyst
Iris cysts are classified into iris stromal cysts and iris pigment epithelial (IPE) cysts.17,18 IPE cysts are far more common than stromal cysts and often simulate melanoma due to their dark brown color. In general, they arise from the posterior surface of the iris, unlike iris melanoma or nevus (Fig. 3). IPE cysts tend to permit some transmission of light on transillumination and appear cystic on UBM. Management is observation unless secondary angle closure occurs due to large size or the cyst enlarges to fill the visual axis.18
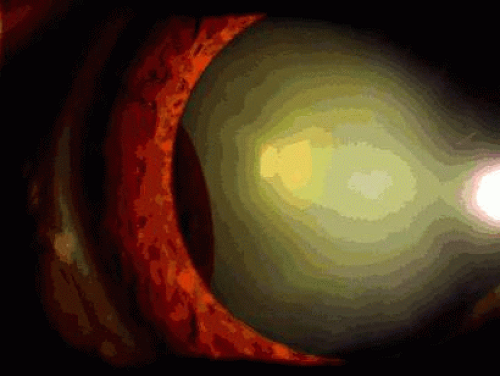 Fig. 3. Iris pigment epithelial cyst (midzonal). |
Iris Pigment Epithelium Epithelioma (Adenoma)
Epithelioma of the IPE is a benign neoplasm that arises from the posterior surface of the iris in the IPE.19,20 Histopathologically, it is composed of columns or acini of pigmented epithelial cells. Clinically it appears as a dark black lesion found in the pupillary space behind the iris or in the angle as a rounded or multinodular mass eroded through the iris stroma (Fig. 4). The lesion typically remains stable or progresses very slowly. Transformation into malignant epithelioma (adenocarcinoma) is exceedingly rare.
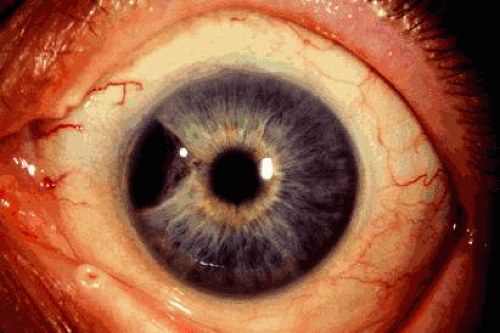 Fig. 4. Epithelioma (adenoma) of iris pigment epithelium thinning the overlying iris stroma. |
The diagnosis of epithelioma of the IPE is best made by clinical recognition of the characteristic features. Tumor extent and configuration is delineated on UBM. Small asymptomatic tumors can be managed by observation. If the lesion shows progressive enlargement or early secondary glaucoma, removal by iridectomy or iridocyclectomy is warranted.
MELANOCYTIC TUMORS OF THE POSTERIOR UVEA (CILIARY BODY AND CHOROID)
Melanocytic tumors of the posterior uvea (ciliary body and choroid) include nevus and malignant melanoma.7 These tumors arise from the uveal melanocytes that are distributed throughout the uveal tract.
Choroidal Nevus
A choroidal nevus is a benign melanocytic tumor that arises in the choroid.21 Although most choroidal nevi remain clinically stationary for many years, they can occasionally give rise to malignant melanoma. It has been estimated that 1 in 5,000 choroidal nevi grow into choroidal melanoma.1 Histopathologically, a choroidal nevus is usually composed of low-grade cells with spindle configuration. Occasionally it is composed of deeply pigmented, round cells with prominent cytoplasm and small nuclei consistent with benign melanocytoma. Choroidal melanocytomas is a variant of nevus that appears clinically as a dark brown lesion, often with a corrugated surface.
Ophthalmoscopically, choroidal nevus is a variably pigmented, well-circumscribed lesion typically found in the posterior choroid. It displays a gray to brown color and is fairly homogeneous (Fig. 5). Hard or soft drusen can be present on the surface of the lesion and generally indicate chronicity of the lesion. Other findings of chronicity include retinal pigment epithelial alterations, fibrous metaplasia, and osseous metaplasia. Pigment epithelial detachment can occur overlying choroidal nevus. Occasionally an choroidal nevus can be amelanotic and resemble choroidal metastasis, lymphoma, and scleritis. Choroidal nevi are usually less than 2 mm in thickness. Accumulation of a minor amount of subretinal fluid can occur related to incompetence of the overlying retinal pigment epithelium (RPE) or, rarely, choroidal neovascularization.
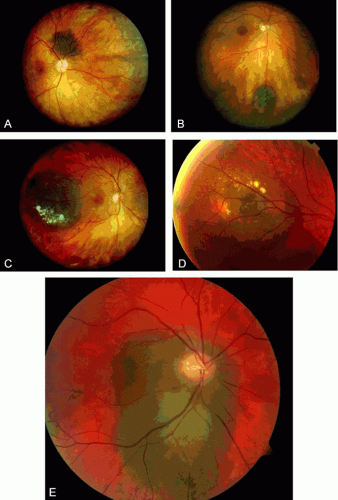 Fig. 5. Spectrum of choroidal nevus. A: Flat choroidal nevus with overlying fine drusen. B: Minimally elevated choroidal nevus with chronic overlying fibrous metaplasia of the retinal pigment epithelium. C: Large choroidal nevus with overlying chronic soft drusen and retinal pigment epithelial atrophy. D: Soft drusen and subtle retinal pigment epithelial atrophy overlying a choroidal nevus. E: Suspicious choroidal nevus encircling the optic disc that has remained stable on 2-year follow-up. |
Visual impairment from choroidal nevus can be related to induced hyperopia from the mass, subretinal fluid, overlying cystoid and noncystoid retinal edema, and overlying photoreceptor degeneration. The latter findings are commonly found on optical coherence tomography.22
Choroidal nevus is best diagnosed by careful ophthalmoscopy by an experienced clinician, recognizing the typical clinical features. Fluorescein angiography may help to differentiate the lesion from subretinal hemorrhage or from lesions of the RPE. The best management of a choroidal nevus is periodic ophthalmoscopy, looking closely for enlargement of the lesion. Serial fundus photography is desirable in order to be certain of any growth. If growth is documented, then the lesion should be managed as a small melanoma. When a nevus produces a secondary retinal detachment that causes visual impairment, then laser photocoagulation or transpupillary thermotherapy to induce resolution of the subretinal fluid is indicated.23
Monitoring choroidal nevi for evolution into melanoma is important. Detection of choroidal melanoma is critical for best patient prognosis. Risk factors have been identified to assist in selecting those tumors at highest risk for transformation into melanoma. These factors include thickness over 2 mm, subretinal fluid, symptoms, orange pigment, and tumor margined within 3 mm of the optic disc24,25,26 (Table 2). The mnemonic of “TFSOM” representing “To Find Small Ocular Melanoma” is useful in remembering the initials of each risk factor. The combination of risk factors imparts greater risk for transformation (Table 3).
TABLE 2. Clinical Features Predictive of Growth of Small Choroidal Melanoma (≤3 mm thickness) | |||||||
|---|---|---|---|---|---|---|---|
|
TABLE 3. Specific Combination of Clinical Features Predictive of Growth of Small Choroidal Melanoma | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ciliary Body Melanoma
A ciliary body melanoma is a malignant neoplasm that arises from the melanocytes of the ciliary body.27 Like iris melanoma, it can contain spindle or epithelioid cells. Because of the hidden location behind the iris, ciliary body melanoma typically attains a large size before clinical detection. External ocular signs, such as dilated episcleral “sentinel” vessels (Fig. 6) or a focus of transscleral extension of the tumor can alert the clinician to a possible underlying neoplasm. Frequently, the tumor grows anteriorly through the iris root and appears in the anterior chamber angle. More typically, however, it is confined to the ciliary body and can only be detected by slit lamp or fundus examination after wide dilation of the pupil Ciliary body melanoma can be uniformly pigmented or partially pigmented, or it can be completely amelanotic.
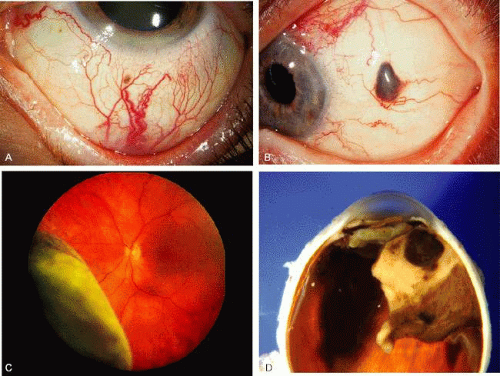 Fig. 6. Features of ciliary body melanoma. A: Episcleral dilated vessels over a ciliary body melanoma. B: Extrascleral extension of ciliary body melanoma. C: Ciliary body melanoma with serous retinal detachment. D: Gross pathology of pigmented ciliary body melanoma. |
Ciliary body melanoma is best diagnosed by recognition of the typical features of the tumor with slit lamp biomicroscopy of indirect ophthalmoscopically through the widely dilated pupil. If the tumor is of sufficient size, ultrasonography can show the typical features of a uveal melanoma. In cases where the diagnosis is challenging, fine-needle aspiration biopsy is employed.13
Choroidal Melanoma
A choroidal melanoma is a malignant melanocytic tumor that arises from the melanocytes within the choroid.21 Like ciliary body melanoma, it contains spindle cells, epithelioid cells, or a combination of the two (mixed cell type). Meta-analysis of published reports has clarified the typical host and environmental features most predisposed to uveal melanoma. Patients with fair complexion, blue or green irides, and freckling are at risk.30 Those who are exposed to arc welding and those with chronic ultraviolet sunlight exposure are also at risk for uveal melanoma.31
The life prognosis for patients with choroidal melanoma depends on numerous factors, one of which is tumor thickness. Melanomas classified as small size (<3 mm thickness) develop metastasis in 16% cases, those that are medium size (3 to 8 mm thickness) show metastasis in 32%, and those classified as large (>8 mm thickness) develop metastasis in 53% of cases32 (Table 4). Very long follow-up of patients with uveal melanoma has found that overall, 60% of patients develop metastasis by 20 to 30 years.33 The greater the thickness, the greater the risk for metastatic disease; hence, there is strong emphasis for early detection of this malignancy.26
TABLE 4. Prognosis of Uveal Melanoma Depends Primarily on Tumor Thickness | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||
In its earliest stages a choroidal melanoma can be clinically indistinguishable from a large benign choroidal nevus. A small choroidal melanoma characteristically occurs as a brown or gray choroidal mass with fairly well-defined borders. Occasionally, orange pigment on the surface of the tumor or subretinal fluid is detected (Figs. 7 and 8). This correlates with clumps of macrophages at the level of the RPE that contain abundant lipofuscin pigment. A larger melanoma usually assumes a dome shape (Fig. 9). A melanoma that is clinically amelanotic is characterized by visible large blood vessels in the substance of the tumor. A choroidal melanoma can eventually break through the Bruch membrane, assuming a mushroom-shaped configuration.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


