Topical Diagnosis: The Optic Chiasm
Joel S. Glaser
(Revised and Updated by Christopher C. Glisson)
The modern neurosurgeon has come to deal largely with lesions that mechanically affect the central apparatus of vision, while the ophthalmic surgeon limits his operative procedure to the intra-orbital portion of the apparatus. One in a sense is extracranial, the other an intracranial ophthalmic surgeon, neither of them venturing to trespass much beyond the narrows of the optic foramina.
Harvey Cushing (1930)
The optic chiasm is the crossroads of the visual sensory system, containing some 2.4 million afferent axons, and it also represents the conjunction of at least four major medical disciplines: neurosurgery, neurology, endocrinology, ophthalmology (and of course, neuro-ophthalmology). Many of the disease processes that involve the intracranial portions of the optic nerves likewise involve the chiasm. Because of the relationship of the nerves and chiasm with the basal structures of the anterior and middle cranial fossae, pituitary adenomas, meningiomas, and aneurysms frequently encroach on the anterior visual pathways. Failure of early diagnosis in chiasmal disorders endangers the life of the patient and lessens the likelihood of reversal of visual and hormonal deficits.
The anatomy of the chiasm is addressed elsewhere in the text, but the following points deserve emphasis in the context of topical diagnosis. The chiasm is situated in the suprasellar arachnoidal cistern, and forms the floor of the anteroinferior midline recess of the third ventricle. The inferior aspect of the chiasm is usually 8 to 13 mm above the nasotubercu-lum line (i.e., the plane of the diaphragma sellae or clinoid processes). The intracranial portion of the optic nerves is inclined as much as 45° from the horizontal, and measures 17 ± 2.5 mm in length. The lateral aspects of the chiasm approximate the supraclinoid portions of the internal carotid arteries, and the anterior cerebral arteries pass over the dorsal surface of the optic nerves as they converge. The optic nerves are fixed at the intracranial exit from the optic canals, the dorsal aspect of which is formed by an unyielding falciform fold of dura.
On the basis of these anatomic relationships, it should be noted that mass lesions, even of moderate size, do not necessarily compress the chiasm. For example, pituitary adenomas must extend well above the sella turcica in order to contact the chiasm. Therefore, if a chiasmal visual field defect is identified, advanced suprasellar extension of an adenoma is most likely present. Small tumors are detected clinically only when signs of unilateral optic nerve compression occur, or endocrinologic symptoms become apparent (in the case of hormonally active lesions).
CLINICAL MANIFESTATIONS
Most chiasmal syndromes are caused by extrinsic tumors (most commonly pituitary adenomas, suprasellar meningiomas, and craniopharyngiomas) or by carotid aneurysms. With few exceptions, these slow-growing tumors produce insidiously progressive visual deficits in the form of variations on a bitemporal theme (Fig. 6.1). The degree of vision loss is often asymmetric; one eye may demonstrate reduced visual acuity and a significant visual field defect, while the visual field of the contralateral eye may show only a slight temporal depression. Until the offending process progresses sufficiently to cause a decrease in visual acuity, the patient’s visual symptoms may be vague (such as “trouble seeing to the side”).
Interesting sensory phenomena are occasionally experienced by some patients with well-developed field defects, consisting of a nonparetic form of diplopia (the so-called “hemifield slide” phenomenon). This can manifest as clumsiness in visual tasks requiring depth perception (e.g., seating the tip of a screwdriver, threading a needle). The loss of portions of normally superimposed binocular field results in the absence of corresponding points in visual space (or on the retinas), and therefore diminished fusional capacity. In essence, the patient is subjected to two “free-floating” nasal hemifields without interhemispheric linkage to keep them aligned. Vertical and horizontal slippage produces
doubling of images, gaps in otherwise continuous visual panorama, and steps in horizontal lines.
doubling of images, gaps in otherwise continuous visual panorama, and steps in horizontal lines.
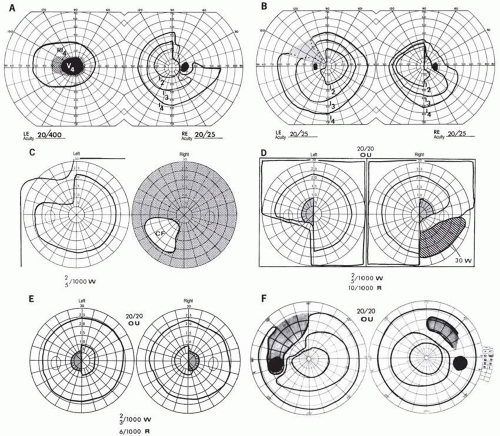 FIG. 6.1 Chiasmal field defects. A: “Junctional scotoma” combines typical optic nerve defect in the left field (LE) with temporal hemianopia in the right (RE) (see also C). B: Classic bitemporal hemianopia. Riddoch’s phenomenon (motion perception) is demonstrable in the shaded area of the left field. C: Asymmetric progression, with severe visual deficit in the right eye and early superior temporal depression on the left. D: Defect characteristic of posterior chiasmal notch lesion. Note the central hemianopic scotomas and inferior quadrant defects. E: Central hemianopic scotomas typical of posterior chiasmal interference. F: Temporal hemianopic arcuate scotomas. The patient sustained head trauma, with resultant field defects and diabetes insipidus. |
The association of extraocular muscle palsies with chiasmal field defects implies involvement of the structures in the cavernous sinus, usually a sign of rapid expansion of a pituitary adenoma. Except in children, it is rare for the diagnosis of a mass lesion to be delayed sufficiently for obstruction of the ventricular system to occur, causing resultant elevation of intracranial pressure and lateral rectus weakness.
Pallor of the optic discs is often anticipated as a logical consequence of chiasmal compression, but is not a requisite in diagnosis. In a series of 156 cases of pituitary tumors, Chamlin and associates1 found optic atrophy in only 155 of 312 eyes (50%). Wilson and Falconer2 also found unequivocal disc pallor in only 28 of 50 patients; they pointed out that optic atrophy may not be present even when visual symptoms have been present for as long as 2 years. Even extensive field loss in chiasmal syndromes may be associated with normal or minimally pale discs. The emergence of optical coherence tomography (OCT) has allowed for objective and reproducible measurement of retinal nerve fiber layer (RNFL) thickness, and thus
a potential strategy for predicting visual recovery following treatment of chiasmal lesions.3, 4 However, based on the slow emergence of optic atrophy over time, greater diagnostic and therapeutic emphasis should be placed on careful evaluation of visual fields. Moreover, improvement of vision to surprisingly good levels in spite of advanced optic atrophy does indeed occur.
a potential strategy for predicting visual recovery following treatment of chiasmal lesions.3, 4 However, based on the slow emergence of optic atrophy over time, greater diagnostic and therapeutic emphasis should be placed on careful evaluation of visual fields. Moreover, improvement of vision to surprisingly good levels in spite of advanced optic atrophy does indeed occur.
Techniques for the identification and characterization of visual field defects have been extensively described elsewhere. Confirmation that the visual field defect is bordered centrally by the vertical meridian is helpful in establishing a chiasmal etiology, and reducing the likelihood of another mechanism of temporal visual field depression. Other conditions that can mimic chiasmal visual field defects include the following:
Tilted discs (inferior crescents, nasal fundus ectasia)
Nasal sector retinitis pigmentosa
Bilateral cecocentral scotomas
Papilledema with greatly enlarged blind spots
Overhanging redundant upper lid tissue
Dominant optic atrophy
Ethambutol toxic optic neuropathy
Central and cecocentral scotomas are usually compelling evidence of intrinsic optic nerve disease. However, cases of “atypical” bilateral scotomas attributed to suprasellar mass lesions have been reported.5 The anterior knee of Wilbrand (a forward looping of decussating fibers 1 to 2 mm into the contralateral optic nerve) may be a pathological artifact,6 but does provide a theoretical basis for the observed pattern of central depression in one eye with contralateral superior temporal hemianopic depression (see Fig. 6.1A).
There are well-documented examples of “posterior chiasmal angle” lesions that produce homonymous hemianopic defects with selective depression of the inferior temporal field near fixation (see Fig. 6.1D). For example, two presumed dysgerminomas and a presumed craniopharyngioma caused these posterior chiasmal defects, at the junction with one optic tract.7 Such patients, unlike posterior pathway hemianopias, have diminished visual acuity in one or both eyes, and asymmetric optic atrophy is usually observed.
NEUROIMAGING PROCEDURES: GENERAL CONSIDERATIONS
Timely completion of necessary radiographic procedures is essential for accurate and outcome-altering diagnosis. The imaging study requested must be appropriate and complete to identify a lesion within the anatomic region of interest. It is imperative that the physician make an informed topical diagnosis based on the clinical findings, and then ask two questions: (1) What studies are appropriate to visualize the relevant anatomy? (2) Are the images obtained (computed tomography [CT], magnetic resonance imaging [MRI], angiography, ultrasonography) of sufficient quality to provide definitive information? On occasion, otherwise informative investigations will be falsely interpreted as “normal,” further delaying definitive treatment. Therefore, it is recommended that radiographic studies, whenever possible, be reviewed by the ordering clinician and the interpreting radiologist in concert.
Specific details concerning neuroimaging techniques are beyond the scope of this chapter, but certain concepts are worthy of note (more detailed clarifications are included with the upcoming discussions of specific lesions).
As a rule, CT provides greater definition than MRI for bone destruction or erosion (e.g., craniopharyngioma, meningioma) and hyperostosis (e.g., meningioma), as well as for vascular or tumoral calcifications (e.g., craniopharyngioma) or acute hemorrhage. MRI more clearly demonstrates vascular encasement (e.g., meningioma), extent of involvement of structures adjacent to neoplasms, invasion of the cavernous sinus, presence of aneurysms, configuration of gliomas, degree of expansion into the suprasellar cistern, central nervous system (CNS) infarcts, and demyelinating lesions. Thin-section MRI is superior to enhanced CT in resolving large pituitary adenomas. The intracanalicular portion of the optic nerve is seen to best advantage with MRI, but bony changes are exquisitely delineated by CT (using “bone-window” protocols). The use of gadolinium-enhanced MRI, with fat-suppression protocols, is especially appropriate for orbital studies. Cerebral arteriography may be included, especially when radiologic, and, in some instances, surgical intervention is contemplated. MRI is relatively contraindicated in the presence of cerebral aneurysm clips, cardiac pacemakers, or ferromagnetic foreign bodies.
High-resolution MRI with thin-section protocols can provide accurate coronal measurement of the intracranial optic nerves (height, 3.5 ± 0.06 mm; width, 6.01 ± 0.1 mm)8 and chiasm (height, 2.69 ± 0.08 mm; width, 14.96 ± 0.33 mm; area, 43.7 ± 5.21 mm2).8, 9 The dimensions of the optic chiasm are reduced by optic atrophy as well as advancing age.8, 9 and 10
CONGENITAL CHIASMAL DYSPLASIAS
Structural differences of the optic chiasm may infrequently occur in the context of developmental anomalies that are related to malformations of other diencephalic midline structures, including the third ventricle. Specific
congenital anomalies of the optic discs may indicate the presence of forebrain malformations that are otherwise not clinically apparent. Optic disc hypoplasia and colo-bomatous dysplasias, therefore, should not be regarded as isolated findings, but prompt appropriate evaluations for potentially associated brain and endocrine defects.
congenital anomalies of the optic discs may indicate the presence of forebrain malformations that are otherwise not clinically apparent. Optic disc hypoplasia and colo-bomatous dysplasias, therefore, should not be regarded as isolated findings, but prompt appropriate evaluations for potentially associated brain and endocrine defects.
NEOPLASMS AND RELATED CONDITIONS
Most cases of chiasm-related vision loss are due to structural disruption of the visual pathways, as a consequence of either tumor or aneurysm. The diagnostic evaluation of all nontraumatic chiasmal syndromes should be focused on the identification of a potentially treatable mass lesion.
Pituitary Tumors
Tumors of the pituitary gland are the single most common intracranial neoplasm resulting in neuro-ophthalmologic symptomatology. Chiasmal interference is overwhelmingly the most frequent presentation. In this setting, pituitary adenomas constitute 12% to 15% of clinically symptomatic intracranial tumors.
Adenomas
Kernohan and Sayre11 reported that asymptomatic adenomas occur in more than 20% of pituitary glands examined at autopsy, and some degree of adenomatous hyperplasia can be found in almost every pituitary gland. A postmortem study12 composed of pituitary glands removed from 120 persons without clinical evidence of pituitary tumors revealed a 27% incidence of microadenomas, of which 41% stained for prolactin (PRL); there was no appreciable gender difference. To generalize, more than 1 in 10 persons in the general population dies harboring a prolactinoma. Moreover, approximately 15% of microadenomas (defined as smaller than 10 mm in diameter, confined within the sella, and frequently disclosed as incidental findings) become significantly enlarged.13 It is not surprising, therefore, that this clinical syndrome is frequently encountered.
Symptomatic adenomas occur infrequently under the age of 20, but they are common from the fourth through the seventh decades. Adenomas have been classified according to their hormonal activity,14 and more recently classification on the basis of morphologic features and immunohistochemistry has proven to be clinically useful.15 Endocrine-inactive tumors do not result in clinical manifestations of any secretory product when: (1) a normal hormone is produced in amounts too small to be detected; (2) when an abnormal hormone is produced but not recognized by biologic receptor sites or detected by radioimmunoassay; or (3) when formerly endocrine-active cells have lost the ability to produce hormone as a result of degeneration.
Nonvisual symptoms of pituitary lesions may include chronic headache (mild or severe), fatigue, erectile dysfunction or amenorrhea, changes in pubic hair, or other signs of gonadal, thyroidal, or adrenal insufficiency. The typical sequence of hormonal deficiencies associated with large adenomas is early loss of growth hormone (GH) and gonadotropin, followed later by loss of thyrotropin and corticotropin. Through the common application of screening neuroimaging studies and sensitive assays for hormonal derangement, the incidence of ophthalmologic presentation is decreasing, whereas the incidence of neuroendocrine findings is increasing. Despite this, signs and symptoms (visual or otherwise) may exist for months to years before a formal visual field test suggests accurate localization of the underlying lesion.2
The patterns of visual field loss occurring as a consequence of pituitary lesions are limited. As suprasellar extension progresses, a single optic nerve may be compromised with worsening monocular visual loss in the form of a central scotoma. More frequently, as the tumor affects the chiasmal fibers, superotemporal peripheral hemianopic defects occur. The commonly recognized and “classic” superior bitemporal hemianopia is almost always accompanied by minor or major hemianopic scotomas approaching the fixational area along the vertical meridian (see Fig. 6.1). Marked asymmetry of the bitemporal defects is not uncommon, such that one eye may be nearly blind while the other shows a temporal hemianopic defect (see Fig. 6.1C); this combination is as exquisitely localizing to the chiasm as is classic bitemporal hemianopia. Adenomas extending posteriorly produce incongruous hemianopias by optic tract involvement; central vision is usually diminished (at least in the ipsilateral eye) and optic atrophy evolves. From a study16 of 50 cases of pituitary adenomas with chiasmal syndrome, it was concluded that visual disturbance does not occur until the chiasm is displaced approximately 10 mm upward.
On extremely rare occasions, the so-called arcuate Bjerrum scotoma extends from the blind spot into the nasal field17 or terminates at the vertical meridian.18 Such defects are usually monocular and may be difficult to distinguish from glaucoma by perimetry alone. With progression, especially if the temporal field of the other eye becomes involved, a more typical field pattern evolves. In late stages of visual loss, the only suggestion of the chiasmal character of field defects may be minimal preservation of the nasal field of one eye (see Fig. 6.1C). Neuroimaging is helpful in determining
the extent of the underlying lesion, but the importance of serial visual field analysis cannot be overstated.
the extent of the underlying lesion, but the importance of serial visual field analysis cannot be overstated.
Management of Pituitary Adenomas
The management of pituitary adenomas should involve several disciplines: (1) neuroimaging with radiology consultation for the detection of microadenomas and to allow precise delineation of gross morphology, signal characteristics, and the status of neighboring structures (Fig. 6.2); (2) radioimmunoassay techniques to assay PRL and other endocrine levels; (3) oral neuropharmacologic agents to provide a “medical adenomectomy” for hyperprolactinemia and acromegaly; (4) transsphenoidal surgery that has all but replaced transcranial approaches; and (5) immunohistochemistry techniques as the “gold standard” of functional classification.15
Prolactin-secreting adenomas (“prolactinomas”) are the single most common type of pituitary tumor, and occur more frequently in women than in men. In women, amenorrhea and galactorrhea are the symptoms that most often prompt medical investigation. In men, symptoms often include loss of libido, erectile dysfunction, gynecomastia, galactorrhea, and hypopituitarism.19 These various clinical manifestations can occur with or without visual loss, depending on the degree of suprasellar extension and compression of the chiasm. As a rule, true prolactinomas are associated with serum PRL levels higher than 150 to 200 ng per mL,
and they usually range from 700 to 7,000 ng per mL. The larger the tumor, the greater the serum PRL. Therefore, ra-diologically large adenomas with PRL levels lower than 200 ng per mL are most likely nonsecreting, and are not likely to respond to neuropharmacologic therapy.
and they usually range from 700 to 7,000 ng per mL. The larger the tumor, the greater the serum PRL. Therefore, ra-diologically large adenomas with PRL levels lower than 200 ng per mL are most likely nonsecreting, and are not likely to respond to neuropharmacologic therapy.
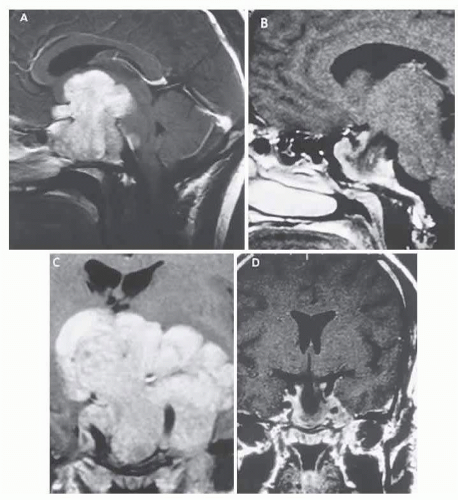 FIG. 6.2 Large prolactinoma. Original vision in the right eye (RE) was 8/200, left eye (LE) 1/200, with serum prolactin of 26,000 ng per mL and galactorrhea. Four months of bromocriptine reduced prolactin to 661 ng per mL, vision improved to RE 20/40, LE 20/50. At 3 years, vision was as follows: RE 20/30, LE 20/20; prolactin was 25.9 ng per mL. Enhanced MRI. Sagittal (A) and coronal (C) images at diagnosis. Sagittal (B) and coronal (D) images at 2-year follow-up, showing dramatic shrinkage of the mass. |
Hyperprolactinemia up to 100 ng per mL may be due to simple physiologic causes including stress, sexual intercourse, nipple stimulation, and exercise, or it may be secondary to pharmacologic agents such as phenothiazines, tricyclic antidepressants, calcium channel blockers, and cimetidine.19 However, other lesions in and around the pituitary gland and hypothalamus that compromise the pituitary stalk may present as “pseudoprolactinomas.” Suprasellar cystic lesions can also cause hyperprolactinemia with field defects,20 as can carotid suprasellar aneurysms.21 Unlike true prolactinomas, non-PRL-secreting suprasellar tumors with secondary hyperprolactinemia due to pituitary stalk compression do not show a correlation between size and PRL level.22
The ergot-derived dopamine agonist bromocriptine provides a pharmacologic alternative (or adjunct) to surgery for prolactinomas. Bromocriptine (2-bromo-α-ergocryptine) is representative of a class of ergot derivatives that inhibit pituitary gonadotropic function, reduce PRL secretion, and diminish the size of pituitary tumors (see Fig. 6.2). Such ergot derivatives are structurally related to dopamine, a PRL-inhibitory factor elaborated by hypothalamic dopaminergic neurons. Bromocriptine decreases PRL production and secretion, with resultant reduction in lactotrope size and subsequent diminution of tumor volume (often rapidly, within 1 to 2 hours of initiation of treatment).19 Spark and associates23 reported the efficacy of bromocriptine in reducing the tumor size; it was demonstrated that bromocriptine lowered PRL, reduced GH in acromegaly, and reversed visual field defects. However, patients with extrasellar extension or with high PRL levels demonstrated a less robust effect. The great weight of evidence now clearly shows that most microadenomas (intrasellar) are demonstrably reduced in size24 in about 3 months at an average dose of 5 mg per day. Cystic necrosis may develop, however, and adenomas may increase in volume if bromocriptine is discontinued.
The clinical course of 10 patients with macroprolacti-nomas at the Wills Eye Hospital in Philadelphia was carefully documented25 after treatment with bromocriptine in daily doses ranging from 7.5 to 30 mg. Nine patients demonstrated rapid improvement in visual acuity and visual fields (often within a few days). There was also a demonstrable decrease in tumor size by CT criteria, and reduction of serum PRL. Four patients subsequently required transsphenoidal decompression for conditions including failure of visual improvement, cerebrospinal fluid (CSF) rhinorrhea, and medication intolerance. It was recommended that patients who are to undergo surgical decompression should be treated preoperatively to decrease the tumor size and “to facilitate surgical removal,” and that residual tumor with elevated PRL should be treated with bromocriptine.
Other dopamine agonists include long-acting bromocriptine (Parlodel) and cabergoline (Dostinex); some prolactinomas resistant to standard dopamine agonists may respond to more potent agents such as cabergoline.26 Ophthalmic results in patients with macroprolactinomas treated with dopamine agonists show generally good results, with few instances of pituitary necrosis.19, 27
The details of neurosurgical treatment of pituitary lesions28 are beyond the scope of the present work. Briefly, within the spectrum of surgical experiences, complications of transsphenoidal procedures may include anterior pituitary insufficiency (about 20%), diabetes insipidus (about 18%), CSF rhinorrhea (about 4%), and, rarely, loss of vision or diplopia.29 Infrequent complications include hydrocephalus secondary to subarachnoid blood, cerebral ischemia related to vasospasm, meningitis with or without CSF leak, and death associated with intraoperative or postoperative hemorrhage.29
Radiation therapy is used for the management of medically and surgically refractory adenomas, or for postoperative treatment in patients with incompletely resected nonfunctional adenomas. External-beam conventional protocols delivering median total dosage of 45 cGy are considered highly effective in preventing recurrence of hormonally inactive tumors, but they may compound relative hypopituitarism.30 Young patients with total tumor removal, or without MRI evidence of recurrence, may be safely observed with radiation therapy held in reserve.
Following uncomplicated surgical decompression, visual acuity and fields may improve rapidly, or gradually. Not all surgical procedures are successful and visual function may worsen, especially after frontal craniotomy for large adenomas with massive suprasellar extension. Visual deterioration at or immediately following surgery is related to intrasellar hematoma formation, edema of tumor remnants, or direct surgical manipulation of optic nerves, chiasm, and the adjacent vasculature. Arterial injuries, for example, may produce intraoperative hemorrhage, delayed epistaxis, carotid arterial occlusion, and pseudoan-eurysm.31 Postoperative packing of the sella with muscle or subcutaneous fat may compress the optic nerves and chiasm.32 MRI is warranted when vision is worsened or does not show signs of improvement within a few days.32
After surgical, medical, or radiation therapy, formal visual field testing should be completed as soon as possible to establish baseline function. In uncomplicated cases, surveillance visual field analysis should occur monthly for the first 3 months, then at 6 months, and subsequently on
a yearly basis. Recurrent visual deficits during the course of monitoring may be caused by regrowth of tumor (most commonly), arachnoidal adhesions associated with a progressive “empty sella syndrome,” or delayed radionecrosis. In the setting of a prolactinoma, serum PRL levels may be monitored; prolactinomas have a higher recurrence rate than nonsecreting tumors.33
a yearly basis. Recurrent visual deficits during the course of monitoring may be caused by regrowth of tumor (most commonly), arachnoidal adhesions associated with a progressive “empty sella syndrome,” or delayed radionecrosis. In the setting of a prolactinoma, serum PRL levels may be monitored; prolactinomas have a higher recurrence rate than nonsecreting tumors.33
Surveillance of treated adenomas can be difficult, particularly with regard to detecting recurrence. As adenomas must be of substantial size to cause visual defects, so must recurrences be large before defects again evolve. Although recurrent vision loss may be the basis for recommending reoperation or irradiation, consecutive perimetry may not reliably reveal “early” tumor recurrence. Therefore, neuroimaging provides the most sensitive technique for monitoring tumor regrowth. Additionally, measurement of serum PRL levels in the immediate postoperative period, and at regular intervals thereafter, is a rational way to monitor for the recurrence of prolactinomas.
Pituitary adenomas may act aggressively on occasion, invading the laterally adjacent cavernous sinuses and producing acute or chronic cranial nerve palsies. Potential immunohistochemical markers for aggressive behavior include p53, MIB-1, PCNA, RB, and H-ras; a high MIB-1 antibody index indicates active proliferation, as does positive p53.34 On rare occasions, prolactinomas may invade neighboring structures.35, 36 Seeding of the subarachnoid space and spread outside the cranium are extremely rare complications that indicate biologic malignancy.
Malignant lesions of the pituitary may be initially mistaken for simple adenomas, including sellar plasmacytoma, lung, and breast metastases37; atypical features suggesting malignancy include rapidly progressive visual loss, ocular motor palsies, and facial numbness. In addition, benign and rare vascular malformations of the sella fossa have been reported.38
Acromegaly
Other adenomas may secrete adrenocorticotropic hormone (ACTH) or thyroid-stimulating hormone or are “mixed” (most commonly PRL- and GH-secreting), but these are principally of endocrinologic interest and are relevant to the ophthalmologist only when extrasellar extension produces visual field defects. However, acromegaly requires further elaboration.
Acromegaly is the clinical condition associated with excess GH, either from autonomous pituitary adenoma secretion or from hypothalamic production of GH-releasing factor with subsequent GH hypersecretion. Many GH-secreting tumors contain a mutant form of the chain of Gs protein in the somatotrope. This represents a relatively rare endocrinopathy, although 228 cases of acromegaly were found among 1,000 adenomas seen at the Mayo Clinic from 1935 to 1972.39 The clinical features include bone and soft-tissue enlargement (especially of hands, feet, and face), visceromegaly, arthritis, carpal tunnel syndrome, hypertension, diabetes, hyperhidrosis, weakness, arthralgias, tooth malocclusion, headaches, erectile dysfunction, menstrual irregularities, and abnormal glucose tolerance. Adenomas associated with acromegaly do not extend beyond the sella as frequently as prolactinomas or nonsecretory tumors, perhaps due to earlier detection as a consequence of prominent clinical manifestations. Nonetheless, 144 of 228 patients with acromegaly in the Mayo Clinic series39 had visual field defects, thus suggesting a delay in diagnosis.
The use of octreotide and other long-acting analogs of somatostatin are indicated for the treatment of patients with active disease when surgery or radiation therapy has failed or is contraindicated; while awaiting the clinical effects of radiation therapy; or as primary treatment in the elderly and medically incapacitated.40 Long-term octreotide therapy reduces serum levels of GH and insulin-like growth factor-1, and it reduces the tumor size.41 Ablation of GH adenomas is also achieved with various forms of radiation therapy, but immediate remission is best accomplished by transsphenoidal resection.
Pituitary “Apoplexy”
Pituitary “apoplexy” refers to an acute change in volume of a pituitary adenoma as a result of spontaneous hemorrhage, edematous swelling, or necrosis. Postpartum infarction or hemorrhage in nontumorous glands does occur, as firmly established in the obstetric literature as “Sheehan syndrome,”42 but chiasmal compression is a rare event. Gross or microscopic hemorrhagic necrosis is independent of endocrine activity or neoplastic pattern. In a review43 of 320 verified adenomas with a high incidence of giant or recurrent large adenomas (41%), evidence of hemorrhage was found in 58 cases (18%). Mean age was 50 years, and the reported clinical course included acute apoplexy (7 cases); subacute apoplexy (11 cases); recent silent hemorrhage (13 cases); and old silent hemorrhage (27 cases). To clarify, in 58 cases of hemorrhagic adenoma, 40 were symptomatically silent. From a series44 of 453 operated adenomas, 45 (10%) demonstrated hemorrhage, but only 13 of these patients had acute symptoms of pituitary apoplexy; the authors correlated hemorrhage with marked suprasellar extension.
Hemorrhage into adenomas has been documented following head trauma,45 after cardiopulmonary bypass,46
and subsequent to tests of pituitary function using thyroid-releasing hormone, gonadotropin-releasing hormone, and insulin.47 Additionally, uncomplicated pregnancy, bleeding disorders, radiation therapy, adrenalectomy, and physical exertion are all reported predisposing factors in pituitary “apoplexy.”48 Moreover, pituitary hemorrhage may occur in adolescence,49 principally in prolactinomas.
and subsequent to tests of pituitary function using thyroid-releasing hormone, gonadotropin-releasing hormone, and insulin.47 Additionally, uncomplicated pregnancy, bleeding disorders, radiation therapy, adrenalectomy, and physical exertion are all reported predisposing factors in pituitary “apoplexy.”48 Moreover, pituitary hemorrhage may occur in adolescence,49 principally in prolactinomas.
The clinical signs and symptoms of pituitary apoplexy include the acute onset of severe headache (often severe frontal or retrobulbar cephalgia or a change in typical headache patterns), acute or rapidly progressing unilateral or bilateral (usually asymmetric) ophthalmoplegia due to rapid expansion into the cavernous sinuses, epistaxis or CSF rhinorrhea when the mass ruptures or erodes into the sphenoid sinus, complications of blood or necrosis debris in the CSF with “pseudomeningitis,” rapid neurologic deterioration and obtundation, and greater or lesser degrees of hypopituitarism.50, 51 Selective expansion laterally into the cavernous sinus may produce ophthalmoplegia without visual loss; selective expansion superiorly may produce visual loss without ophthalmoplegia. Almost without exception, enlargement of the sella is found even on plain skull film views; both CT and MRI detect recent hemorrhage (Fig. 6.3), but MRI may fail to demonstrate acute hemorrhage unless specific sequences are employed (hemorrhage may be isointense on T1 weighted images and hypointense on T2 weighted images; in the subacute phase, extracellular methemoglobin should appear bright on both T1 and T2 sequences). Corticosteroid replacement and other supportive measures may be critical, and, in most instances, immediate decompression through the sphenoid sinus is required. Bromocriptine has been suggested as a temporizing measure when signs and symptoms are modest and not rapidly progressive,52 and there are advocates53 for conservative management consisting of intravenous dexamethasone in the event of minimal or rapidly improving visual deficits. Pituitary apoplexy is often a delayed diagnosis (being confused with ruptured aneurysm or meningitis, for example), and as catastrophic consequences can be mitigated by prompt recognition, it is advisable to review the above symptoms with all patients harboring pituitary lesions and instruct them to seek immediate medical attention in the event that they occur.
Meningiomas
Posterior perioptic foraminal, medial sphenoid ridge, and tuberculum sellae meningiomas, and posteriorly extending olfactory groove and planum masses can produce prechi-asmal (optic nerve) or chiasmal compression. Visual deficits usually take the form of slowly progressive monocular loss of vision, and, when both fields are involved, there is a distinct tendency toward marked asymmetry (frequently with an extensive visual deficit on one side before the contralateral field becomes involved). Slow growth across the tuberculum eventually results in contralateral optic nerve or chiasmal interference. There is a distinct predilection for meningiomas to occur in middle-aged women. Furthermore, enlargement during pregnancy and a possible association with breast cancer are supportive evidence for the role of estrogen and progesterone receptors.54
Although nonspecific headaches are a common feature of suprasellar meningiomas, most patients present with monosymptomatic failure of vision, and are thus likely to present initially to the ophthalmologist. Although relentless deterioration of vision is the rule, fluctuations over weeks or months may mimic optic neuritis.55 In a series of suprasellar meningiomas,56 the time interval from the onset of unilateral visual loss to subjective bilateral defects ranged from 1 to 8 years; simultaneous bilateral onset was not reported. Although periocular pain made worse by eye movement is typical of inflammatory optic neuritis, Ehlers and Malmros57 found that this can occur with suprasellar meningiomas.
Asymmetric optic atrophy is a relatively “late sign,” with normal-appearing optic discs being fully compatible with gradual and progressive visual loss occurring over many months. The much-touted Foster Kennedy syndrome (optic atrophy with contralateral papilledema due to large subfrontal meningiomas) remains a distinct rarity.57 Anosmia, also classically considered an important finding with olfactory meningiomas, is infrequently encountered and may be nonspecific. Delayed diagnosis of large tumors results in frontal lobe compression and edema, potentially causing cognitive changes or hydrocephalus through obstruction of the ventricular system.
Currently applied neuroimaging techniques, including enhanced CT with “bone-window” protocols and gadolinium-contrasted MRI, are exceedingly sensitive in disclosing meningiomas or other parachiasmal masses (Fig. 6.4). At present, contrast-enhanced CT or MRI precisely demonstrate extra-axial tumor configuration; CT is superior in disclosing calcification or bone changes but is inferior for the assessment of suprasellar or intrasellar extension, postsurgical changes, and vascular displacement or encasement.58 Fortunately, clinical and radiographic techniques are sufficient for diagnosis, eliminating the need for vision-threatening tissue biopsy in almost all cases.
Treatment Considerations
In general, growth and regrowth rates for meningiomas are extremely slow, compounding the problem of assessing options for therapeutic intervention. Most advocate careful observation in the absence of intracranial
extension or deleterious effect on vision. In a large surgical series,59 257 patients underwent 338 craniotomies for meningiomas at diverse intracranial locations. Of these, there were 35 sphenoid wing, 20 olfactory groove, 12 tuberculum sellae, and two orbitocranial meningiomas; that is, about 27% of tumors were of ophthalmic interest. For the entire series, the average observed survival was 9 years, and recurrence rate was 22% overall. In a series collected at the Massachusetts General Hospital,60 of 225 patients operated on for meningioma, parasellar tumors constituted 12%. Only half of these were considered grossly “completely excised,” but the 5-year probability of recurrence or progression was reported as 19%.
extension or deleterious effect on vision. In a large surgical series,59 257 patients underwent 338 craniotomies for meningiomas at diverse intracranial locations. Of these, there were 35 sphenoid wing, 20 olfactory groove, 12 tuberculum sellae, and two orbitocranial meningiomas; that is, about 27% of tumors were of ophthalmic interest. For the entire series, the average observed survival was 9 years, and recurrence rate was 22% overall. In a series collected at the Massachusetts General Hospital,60 of 225 patients operated on for meningioma, parasellar tumors constituted 12%. Only half of these were considered grossly “completely excised,” but the 5-year probability of recurrence or progression was reported as 19%.
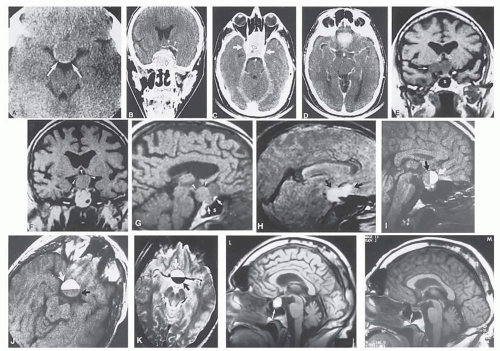 FIG. 6.3 Neuroimaging of pituitary adenomas. A: Axial CT section shows a round tumor mass filling the suprasellar cistern; ring enhancement (arrows) indicates subcapsular hemorrhage. B: Contrast-enhanced coronal CT section through a large invasive adenoma. Note the encasement of the carotid artery (arrows) and the position of the middle cerebral artery above (arrowheads). C: Axial CT section shows lateral expansion into the cavernous sinuses (Black arrows) and a necrotic cyst (black arrow). D: Subfrontal superior extent of the mass. Note the middle cerebral arteries. E: MRI of a large lobulated prolactinoma, with suprasellar extension. Note the distortion of the third ventricle (open arrows) and extension toward the temporal lobe (long arrow); the tumor has not involved the sphenoidal sinus (s). F: Chiasm (arrowheads) is draped on the superior surface of the tumor (TR, 550 milliseconds; TE, 26 milliseconds). G: Sagittal section shows suprasellar growth with the chiasm above (arrowheads); the sella (arrows) and sphenoidal sinus (s) are normal (TR, 850 milliseconds; TE, 26 milliseconds). H: Hyperintense signal (TR, 2000 milliseconds; TE, 60 milliseconds) indicates the partial cystic character (arrows). Sagittal (I) and axial (J) sections with head tilt to the right, in case of a large cystic adenoma with an interface level between newer blood (white arrow) and older blood (black arrow) (TR, 800 milliseconds; TE, 30 milliseconds). K: Signal difference is intensified (TR, 2100 milliseconds; TE, 80 milliseconds) (arrows). L: Hemorrhage (bright signal, arrow) in a pituitary adenoma with headache and bitemporal field depressions. M: Without interventions, 2-month follow-up showed spontaneous involution, with normal pituitary gland (arrow), stalk, and chiasm. |
In a series of 12 incompletely resected meningiomas (8 sphenoidal, 2 petrosal, 1 each orbital and parasagittal), the patients were subjected to radiation therapy of 4,800 to 6,080 cGy (median dose, 5,490 cGy) in 6 weeks61; the median postradiation follow-up was 54.5 months (range, 20 to 120 months), and 9 patients were said to remain free of clinical or radiologic signs of tumor progression. Recurrent lesions were discovered at 4, 6, and 9 years, and the authors concluded that postoperative radiation therapy is
indicated for incompletely excised meningiomas. Carella and colleagues62 reviewed their experience with 68 patients (49 women and 19 men) divided into three groups: (A) 43 patients who had surgery (42 with known residual tumor) followed by irradiation; (B) 14 patients who had radiation for recurrence, of whom 11 underwent subtotal resection before radiation therapy; and (C) 11 patients who had radiation as primary treatment. In Group A, 41 of 43 patients were alive, most doing well neurologically after 1 to 10 years; in Group B, 5 were dead of meningioma (all within 3 years), and 7 patients were considered “stable.” In Group C, all were alive at 3 to 6 years, 9 with neurologic improvement, and 4 with CT evidence of tumor shrinkage with central necrosis.
indicated for incompletely excised meningiomas. Carella and colleagues62 reviewed their experience with 68 patients (49 women and 19 men) divided into three groups: (A) 43 patients who had surgery (42 with known residual tumor) followed by irradiation; (B) 14 patients who had radiation for recurrence, of whom 11 underwent subtotal resection before radiation therapy; and (C) 11 patients who had radiation as primary treatment. In Group A, 41 of 43 patients were alive, most doing well neurologically after 1 to 10 years; in Group B, 5 were dead of meningioma (all within 3 years), and 7 patients were considered “stable.” In Group C, all were alive at 3 to 6 years, 9 with neurologic improvement, and 4 with CT evidence of tumor shrinkage with central necrosis.
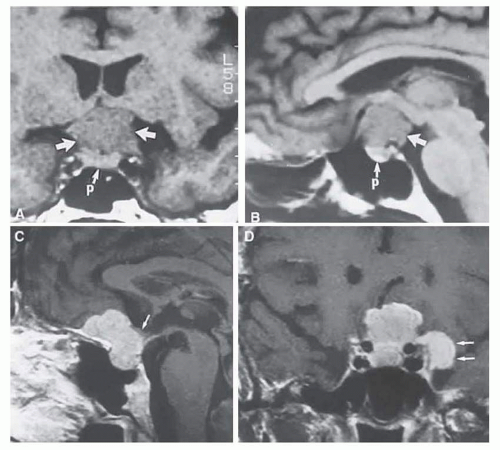 FIG. 6.4 MRI of a suprasellar meningioma (TR, 600 milliseconds; TE, 20 milliseconds). A: Coronal section of a large meningioma (large arrows), isodense to brain. B: Sagittal section. Note the normal sella and pituitary gland (p). Sagittal (C) and coronal (D) sections of a planum meningioma, extending into the sella. Note the upward deflection of the chiasm (arrow in C) and extension to the cavernous sinus (arrows in D). |
Currently, fractionated radiation is regarded as the appropriate therapy for long-term preservation of visual function for nondiabetic patients with primary optic nerve sheath meningiomas (ONSM).63, 64 Surgical resection for meningiomas of the optic nerve sheath is technically complicated, and carries a high risk for postoperative vision loss.63 Therefore, except in atypical or very specific circumstances,65 radiation therapy is preferable.
Chemotherapy (combination therapy with 5-fluorouracil/folate/levamisole or intra-arterial cisplatin and intravenous doxorubicin) is a consideration for patients with recurrent or treatment-unresponsive ONSM. Progesterone receptor sites are expressed in 81% of women and 40% of men with meningiomas,54 with 96% of benign and 40% of malignant meningiomas containing progesterone receptor-positive nuclei, but without correlation between progesterone receptor index and age or histologic subtype.54 Moreover, the efficacy of antiprogesterone agents such as mifepristone has proved disappointing.66 At present, immunotherapy in the form of interferon-α is the most frequently utilized, and is well-tolerated when necessary, for management of optic nerve sheath meningioma.67
Craniopharyngiomas
Craniopharyngiomas are tumors that arise from vestigial epidermoid remnants of Rathke pouch, scattered as cell rests in the infundibulo-hypophyseal region. These tumors are usually admixtures of solid cellular components and variable-sized cysts containing oily composites of degenerated blood and desquamated epithelium or necrotic tissue (and blood) with cholesterol crystals. Dystrophic calcification of this debris is detectable with plain films and CT imaging, and is an important radiologic sign estimated to be seen in more than 80% of childhood craniopharyngiomas. These tumors are congenital (dysontogenic) and, in rare instances, may present in the neonate. There is a more or less bimodal age incidence, peaking in the first two decades and again between 50 and 70 (Fig. 6.5). These predominantly suprasellar tumors account for 2% to 4% of all intracranial tumors regardless of age group, but the incidence is 8% to 13% in children. Of all suprasellar masses, craniopharyngiomas comprise 54% in children and 20% in adults, and show two clinicopathologic and pathogenetic separate types: adamantinous (predominantly cystic, in childhood) and squamous-papillary (predominantly solid, in adulthood).68
The presentation in childhood is commonly related to hydrocephalus and endocrinopathies, consisting of variable degrees of hypopituitarism with or without diabetes insipidus; obesity and somnolence also attest to hypothalamic disturbance. Gonadotropic hormone deficit results in slowed or absent sexual development; precocious puberty is rare. In children, progressive visual loss goes unnoticed until a level of severe bilateral impairment is reached, or unless headache, vomiting, and behavioral changes occur. Increased intracranial pressure produces papilledema in about 65%, and optic atrophy is observed in roughly 60%.69
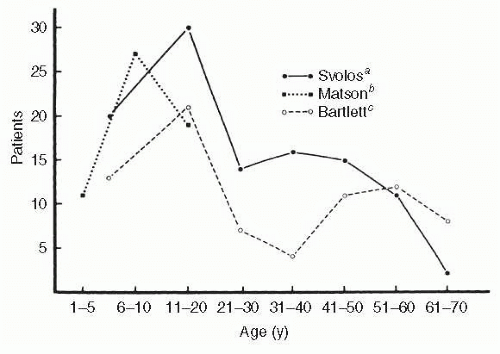 FIG. 6.5 Age distribution of craniopharyngioma. (Data fromaSvolos D. Craniopharyngiomas: a study based on 108 verified cases. Acta Chir Scand Suppl. 1969;403:1;bMatson DD, Crigler JF. Management of craniopharyngioma in childhood. J Neurosurg. 1969;30:377; andcBartlett JR. Craniopharyngiomas: an analysis of some aspects of symptomatology, radiology and histology. Brain. 1971;94:725. The Matson series was limited to children younger than 16 years.) |
In adults, visual deterioration is the universal symptom that demands investigation, although occult endocrine dysfunction may be uncovered. Hypopituitarism, diabetes insipidus, amenorrhea, and galactorrhea inconstantly eventuate. Visual field defects frequently consist of asymmetric bitemporal hemianopias, or a homonymous pattern with reduced acuity (Fig. 6.6) when the optic tract is compressed.70
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


