Topical Diagnosis: Prechiasmal Visual Pathways
Valerie Purvin
Joel S. Glaser
Part I. The Retina
To suppose that the eye, with all its inimitable contrivances for adjusting the focus to different distances, for admitting different amounts of light, and for the correction of spherical and chromatic aberration, could have been formed by natural selection, seems, I freely confess, absurd in the highest possible degree.
Charles Darwin
“Organs of extreme perfection and complication”
In The Origin of Species, 1859
Accurate diagnosis of disorders of the visual sensory system is dependent on knowledgeable history taking and competent evaluation of visual function, including acuity, visual fields, and color perception, of pupillary light reactions and of the fundi. There are no valid diagnostic shortcuts; rather, there are guiding principles and axioms that should at least determine a provisional impression and course of investigation, if not provide a conclusive diagnosis. Part I of this chapter is intended to provide a reasonably detailed overview of those retinal disorders and diseases that may confound or otherwise touch on neuro-ophthalmologic diagnosis.
SYMPTOMATOLOGY
Diseases and pathologic alterations involving the retina provoke the least clinical dilemma in that, for the most part, the ophthalmoscope resolves the question of the anatomic level at which the visual pathways are involved. The problem of complicated neurodiagnostic studies should not arise, although fluorescein angiography, electroretinography (ERG), and other retinal function tests (see Volume 2, Chapter 2) may prove valuable. There are two major pitfalls to be avoided. First, minimal retinal changes may be misconstrued as the cause of disproportionately perturbed visual function, or a normal macula may be misinterpreted as abnormal. For example, a patient with relentless monocular visual loss, a central field depression, and afferent-defect pupil, with a few drusen or minimal derangement of retinal pigment epithelium at the fovea, must not be dismissed with an inappropriate diagnosis of “macular degeneration.” In this instance, the afferent-defect pupil indicates a conductive lesion of the optic nerve and cannot be attributed to minimal retinochoroidal disease. Second, true macular disease, especially when bilateral and ophthalmoscopically subtle, should raise the question of macular dystrophies that masquerade as neurologic disease or for which no cause is apparent (Fig. 1). Indeed, there are increasing numbers of retinal disorders that produce subtle or even insignificant objective fundal changes that may escape conventional ophthalmoscopic detection.
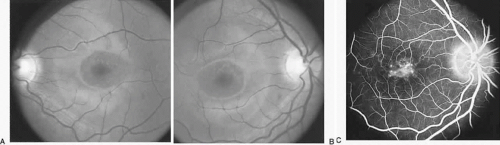 Fig. 1. A 14-year-old boy referred for occult neurologic disease or malingering who had undergone magnetic resonance imaging and psychiatric counseling. Visual acuity was 20/100 in both eyes. Fundi (A and B) show thinned rounded macular reflexes and mild pigment changes at the foveae. Fluorescein angiography (C) disclosed marked macular pigment epithelial disturbance. Diagnosis: juvenile macular degeneration (Stargardt’s type). |
These “hidden” retinal disorders include the following: congenital and hereditary photoreceptor or pigment epithelial abiotrophies and dystrophies; immune-mediated retinopathy associated with distant carcinoma; and an enlarging list of acute zonal occult outer retinopathies, conveniently labeled AZOOR by Gass and associates.1,2 Previously lumped as the “big blind spot syndrome,” AZOOR now encompasses a variety of heterogeneous, presumably inflammatory, retinopathies to be discussed below. Other lesions such as serous detachment of the macula (central serous choroidopathy) or cone dystrophies may be quite subtle on funduscopic examination alone, even during biomicroscopy with the use of a corneal contact lens or Hruby lens. It is in such situations that auxiliary tests of visual function, including color function, Amsler grid, photostress, ERG, and fluorescein angiography are critical in distinguishing modest lesions involving the choroid and retina from early optic nerve compression and demyelination.
In the assessment of visual function, the role of retinal aging alone is noteworthy. Even as macular photoreceptors are incompletely developed at birth and do not reach maturity until at least 4 years post partum, so virtually every parameter of visual function declines from mid-adulthood onward. Age-related degradations in reading acuity, color perception, and feature recognition (contrast sensitivity) are caused by senescent changes of neuronal elements, and, in fact, over a 70-year life span there is a loss of almost 50% of retinal ganglion cells, some half of which serve the macula.3 After the age of 40 years, there is apparently a linear net loss of cone photoreceptors especially from the fovea, to which is added the non-neural factors of increasing pupillary miosis and decreasing lens transparency.4 Other studies dispute an age-related reduction in rod and cone counts.5
The temporal profile of monocular visual deficit is often an important clue in differential diagnosis. Abrupt, non-transient visual symptoms usually indicate retinal artery or vein occlusion, infarction of the optic disc, retinal detachment, or hemorrhage into the vitreous. Age-related macular degeneration may suddenly cause hemorrhage beneath the fovea. In addition, on occasion, optic neuritis may run a course that is interpreted by the patient as abrupt, rather than subacute or rapidly progressive. Transient visual events are considered below (see Table 3).
TABLE 3. Transient Obscurations of Vision | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
HEREDODEGENERATIONS AND ABIOTROPHIES
PIGMENTARY RETINOPATHIES
Heredodegenerative retinal disorders such as retinitis pigmentosa are almost always separable from “neurologic” causes of visual deficits on the basis of chronicity, bilaterality, and typical funduscopic appearance of pigmentary retinopathy, attenuated arteriolar tree, and waxy disc pallor. “Retinitis pigmentosa” is actually a misnomer twice over: the progressive photoreceptor degeneration is not inflammatory, as “-itis” would imply; and pigmentary changes evolve relatively late and may not be obvious. The term “retinitis pigmentosa sine pigmento” is not a separate entity, but it represents a stage of disorder when the retina appears relatively unaltered but photoreceptor function is defective.
A thorough account of retinal photoreceptor dystrophies is beyond the scope of neuro-ophthalmologic subjects, but A. C. Bird’s Jackson Lecture6 provides an overview of current concepts of clinical classification, molecular biology, and therapeutic advances. Hereditary transmission may follow autosomal dominant or recessive, or X-linked, patterns, with a spectrum of responsible gene mutations. Indeed, molecular geneticists have now identified numerous mutations in literally dozens of genes that are associated with or cause photoreceptor degeneration, findings suggesting an extraordinary vulnerability of these cells, possible stemming from their high-oxygen-requiring metabolism and physiologic population culling. These detailed genetic elaborations are replacing imprecise, if more classic, clinical descriptions.6
In the fully developed disorder, there is usually profound constriction of the fields with relative sparing of the central fixational area, resulting in so-called “gun-barrel” or “tubular” field constriction. End-stage retinitis pigmentosa is actually one of the few organic causes of markedly narrowed visual fields. The causes of such “gun-barrel” field constrictions are listed in Table 1. The retained field function actually mimics a cone (see Volume 2, Chapter 2, Fig. 19), because the central remnant enlarges as the testing distance is increased between the patient and, for example, the tangent screen. The field diameter does not enlarge with increasing testing distances in hysteria or malingering, and, in fact, the diameter of the field may shrink further if this possibility is suggested to the naive patient.
TABLE 1. Constricted Field with Retained Acuity | ||||||
|---|---|---|---|---|---|---|
|
Atypical cases of selective pigmentary degeneration of the nasal retinal sectors (Fig. 2) produce field defects that may mimic bitemporal hemianopias. Such preferential involvement of the fundus nasal to the optic disc occurs in at least one-third of patients with sectoral retinitis pigmentosa. Nerve fiber bundle scotomas, somewhat mimicking glaucoma, are also recorded.7,8
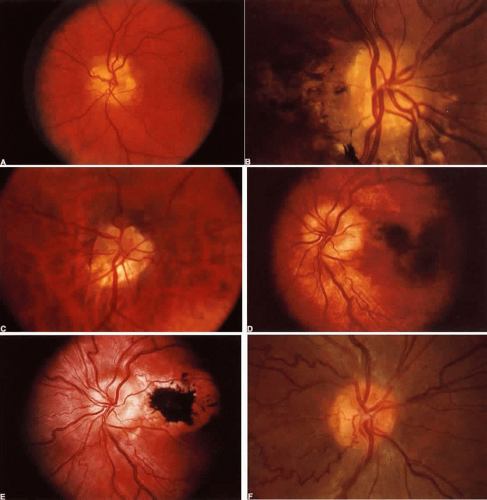 Fig. 2. Retinitis pigmentosa. A. Advanced field loss showing dense annular defects. Deficits start in the 20° to 30° middle zone (as compared with Bjerrum’s zone defects in glaucoma) and proceed toward fixation and outward toward the periphery. Central fixation is relatively spared, producing “gun-barrel fields.” B. The pseudobitemporal field defects of sector retinitis pigmentosa. Unlike chiasmal interference, the defects cross the vertical meridian. C. Left fundus of patient with nasal-sector retinitis pigmentosa. |
Acuity is reduced when cystic or wrinkling changes occur at the fovea or as the central aperture of field finally darkens. Disc pallor and retinal arteriolar attenuation are not simply the result of ganglion cell death because the ganglion cell and nerve fiber layer of the retina are affected only late in the disease. Unilateral or bilateral disc hyaline bodies (drusen), at times most marked in peripapillary and juxtapapillary locations, occur in some 10% of all genetic subtypes.9
Central field defects with acuity loss occur with photoreceptor degeneration that especially affects the macula, so-called retinitis pigmentosa inversa, but this disorder likely represents a separate nosologic class, the cone-rod dystrophies (see below) (Fig. 3). Severe visual defect in early infancy is frequently enough caused by a primary outer segment retinal abiotrophy, Leber’s congenital amaurosis, although this is probably not a single clinical entity. This disorder is characterized by the following: severe impairment of vision, present at birth or becoming evident during early to late infancy; a fundus that may initially approach normal, but within years, optic atrophy, diffuse fine pigmentary degeneration, and attenuation of the arteriolar tree are evident; and either an absent or a markedly reduced ERG response. Other variable features include nystagmus, photophobia, digito-ocular maneuver (forceful eye rubbing with sunken globes; “blindisms”), strabismus, cataracts, hyperopia, mental retardation, deafness, renal anomalies, seizures, hydrocephalus, and focal neurologic deficits (e.g., cerebral diplegia). Hereditary transmission is typically autosomal recessive10 (see Volume 2, Chapter 13).
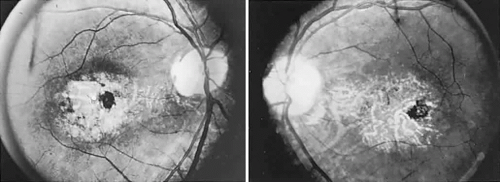 Fig. 3. Cone-rod dystrophy or so-called “retinitis pigmentosa inversa” in a young man with progressive spinocerebellar degeneration and 20/200 acuity in each eye. |
CONE AND CONE-ROD DYSTROPHIES
As noted above, the macular (“inversa”) form of abiotrophic pigmentary retinopathy is actually a selective loss of cone function, but rod function also is defective. Indeed, retinal dystrophies accompanying many systemic disorders are typically, but not exclusively, this macular form. Of special neuro-ophthalmologic interest is the association of this and other geographic pigmentary patterns with hereditary cerebellar ataxias,11,12 with olivopontocerebellar and spinocerebellar degenerations,12,13,14 and with Friedreich’s ataxia.15 These disorders are usually dominantly inherited, with variable expression of early life onset of progressive spasticity, ataxia, slowed saccades or supranuclear ophthalmoplegia, and chorioretinal macular dystrophy. Trinucleotide gene expansion (point mutation) is incriminated, but not exclusively. In Friedreich’s ataxia, the most common inherited, if clinically inhomogeneous, spinocerebellar ataxia, a mitochondrial location of frataxin (Friedreich’s ataxia protein) has been identified; this locus on chromosome 9 reflects the relationship with vitamin E deficiency ataxia and certain neuropathies with mutations in nuclear genes.16
Other subsets of pigmentary retinopathies are due to mitochondrial DNA mutations and are associated with migraine, ataxia, dementia, and Leigh’s disease17; the sporadic or maternally inherited MELAS syndrome of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes, usually presents in the teens as cognitive regression, headaches, and cerebral lesions causing field defects.18,19,20 Even in family members, there is considerable variation of genotypic and phenotypic specificity in metabolic disorders mediated by mitochondrial DNA aberrations, including Kearns-Sayre progressive external ophthalmoplegia (see Volume 2, Chapter 12) and other conglomerations of pigmentary retinopathies. Further identification of gene point mutations will eventually provide a more precise classification.
Progressive cone degenerations present as bilaterally diminished acuity, defective color vision, and aversion to bright lights (photophobia) with “day blindness” (hemeralopia). Central field defects progress, at times showing a fenestrated central scotoma. This widespread loss of cone function usually begins in the first 2 decades of life, but severe cone disease may begin at any age. Heredity is autosomal dominant, but sporadic cases are common, and both severity and rate of progression are variable. The fundus may appear quite normal, a finding that, when coupled with photophobia, provokes an impression of hysteria, but defective pigment epithelium in the form of a “bullseye,” nonspecific mottling, and crystalline deposits are usually enhanced by fluorescein angiography (see also Fig. 1). Mild disc pallor is not unusual. Abnormal single-flash photopic and flicker (i.e., cone-mediated) responses define the disorder by ERG21; fluorescein angiography and full-field ERG may be inconclusive, yet focal ERG is abnormal.22 If considered sooner rather than later, full-field or focal ERG should obviate more exhaustive and inappropriate diagnostic studies for optic nerve disease.
Cones may be congenitally absent, as in the case of rod monochromatism, a rare form of “color blindness” with acuity of about 20/200 and reduced visual capacity under bright light situations; that is, these children show a preference for dim illumination. Pendular or mixed jerk-pendular nystagmus patterns are typical, and the fundus foveal reflex may be blunted. ERG shows absent or markedly subnormal photopic (cone) responses, but normal scotopic (rod) responses.
Pigmentary retinopathies, or more typically cone-rod dystrophies, keep company with a large number of other neurodegenerative and metabolic disorders, such as Bassen-Kornzweig syndrome, Refsum’s disease, Kearn-Sayre progressive ophthalmoplegia (see Volume 2, Chapter 12), Laurence-Moon-Bardet-Biedl cerebellar ataxia, and sensorineural hearing loss. A complete description of the retinal abiotrophies, dystrophies and degenerations may be found in topical reviews, and pertinent texts. Age-related macular degenerations and other macular dystrophies and abiotrophies without neurologic implications are beyond the scope of this chapter.
CHORIO-RETINAL INFLAMMATIONS
As noted above, Gass1 and others have elaborated a class of acute and subacute, diffuse or focal, presumed inflammatory zonal disorders of the outer retinal layers, now termed AZOOR. Roughly described as “enlarged blind spots” and thus confounding neurologic diagnosis, this somewhat heterogeneous rubric includes MEWDS (multiple evanescent white dot syndrome), multifocal choroiditis, acute macular neuroretinopathy, pseudo-presumed histoplasmosis, and idiopathic blind spot enlargement. These entities share a constellation of signs and symptoms and so raise the question of a spectrum of common origin involving geographic zones of retinal photoreceptors and pigment epithelium. Characteristics include the following: predilection for female patients; acute onset in one or both eyes, associated with photopsias; minimal fundus findings at onset, but eventual minor pigment epithelial disturbances; ERG abnormalities; fluorescein angiographic evidence of geographic thinned pigment epithelium; vitreous cells; and permanent field depressions often close to the physiologic blind spot. Taken as a group, Jacobson and colleagues2 investigated the nature of retinal dysfunction and found patchy but dense scotomas and ERG abnormalities, but no evidence of autoantibodies to specific retinal antigens. Recurrent central nervous system (CNS) inflammation in association with AZOOR is reported,23 characterized by cerebrospinal fluid (CSF) lymphocytosis and multiple magnetic resonance imaging (MRI) signal abnormalities, followed in 6 years by an episode of cervical myelopathy.
Acute multifocal retinitis, another idiopathic inflammation in eyes of healthy young to middle-aged adults, may be associated with optic disc edema and may follow flulike prodromes.24 Mild vitrous reaction and macular exudative stars are characteristic, and a self-limited benign outcome is usual, with no evidence of specific infectious or autoimmune causes. Other relatively acute multifocal chorioretinal inflammations include presumed ocular histoplasmosis, coccidioidomycosis, Pneumocystis carinii infection, cryptococcosis, mycobacterial or syphilitic choroiditis, birdshot (vitiliginous) retinochoroidopathy, punctate inner choroidopathy, and acute multifocal posterior placoid pigment epitheliopathy (AMPPPE). Although specific infectious agents may also be incriminated in meningeal reactions, such as cryptococcosis, AMPPPE is associated with CSF pleocytosis and protein elevation, findings suggesting possible viremia.25 The foregoing discussion has focused on retinal pigmentary and macular disease in relation to neurologic disorders, not simple isolated macular dystrophies, which are considered at length elsewhere.23,26
METABOLIC STORAGE DISORDERS
Biochemical assays have considerably clarified the nosologic status of the group of storage diseases previously classified as the “cerebromacular degenerations,” of which Tay-Sachs disease is the eponymous prototype. Although these disorders share a superficial resemblance, showing a progressive neurodegenerative course with variable fundus findings, they are now best classified by abnormal storage products (e.g., sphingolipidoses, mucopolysaccharidoses, and mucolipidoses) and lysosomal enzyme deficiencies. Lysosomes contain hydrolytic enzymes that degrade proteins, polysaccharides, and nucleic acids; if undegraded, these materials accumulate in lysosomes and impair cell function. The complex lipids and saccharides indigenous to neural cells produce symptoms and signs related to eye and brain, including corneal clouding, macular “cherry-red spot,” pigment epithelial degenerations, optic atrophy, mental deterioration, seizures, motor incoordination, myoclonus, and death.
The ganglion cell layer of the retina is a principal site of abnormal accumulation of anomalous storage products, such that ophthalmoscopic changes are observable either in the form of retinal “graying” or the well-known cherry-red spot. The ganglion cell layer densely surrounds the thin fovea, which transmits the normal red color of underlying choroid (Fig. 4). The storage disorders with cherry-red spot or macular graying are listed in Table 2.27
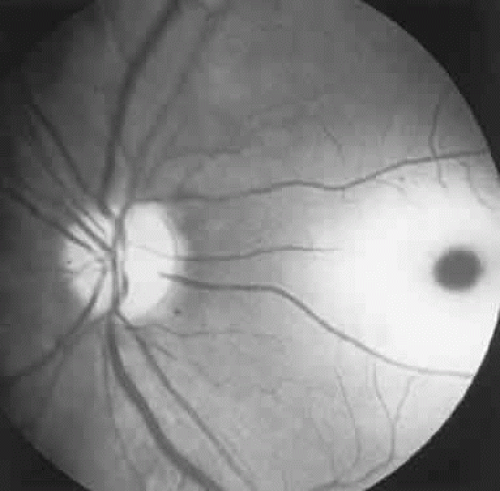 Fig. 4. “Cherry-red spot” of advanced Tay-Sachs disease (gangliosidosis). Note the central foveal window surrounded by a ring of densely opaque retinal ganglion cell layer; also, optic atrophy. |
TABLE 2. Storage Diseases with Macular Changes | |||||
|---|---|---|---|---|---|
|
In addition to retinal changes, pallor of the optic disc occurs in numerous storage disorders, the nerve being affected by several mechanisms. Optic atrophy occurs when abnormal glycolipids are stored in retinal ganglion cells with subsequent neuronal death and nerve pallor, such as occurs in Tay-Sachs disease (Gm2 gangliosidosis), but ophthalmoscopic evidence of optic atrophy is not invariable. Tay-Sachs is an autosomal recessive inherited disease, resulting from mutations of the hexosaminidase (Hex) A gene coding for the alpha-subunit of beta-D-N-acetyl-hexosaminodase. Juvenile and adult Hex A deficiencies are rare, less severe variants. Both infantile and late forms are most prevalent among Ashkenazi (roughly, Middle and Eastern European) Jews, but are reported in non-Jews, in whom clinically benign mutations may occur.28,29 MRI findings include hyperintensities in basal ganglia and thalamus, with marked brain atrophy and diffuse white matter lesions; these findings likely reflect accumulation of Gm2 ganglioside.30 Similarly, in Gm1 gangliosidosis (generalized gangliosidosis; “Tay Sachs disease with visceral involvement”), there is also an abnormal deposition of ganglioside in the retina, with subsequent atrophy of the nerve, but it is a mucopolysaccharide that accumulates in the viscera.
Metachromatic leukodystrophy (sulfide lipidosis) is an autosomal recessive deficiency of arylsulfatase A, with ceramide-galactose sulfate in retinal ganglion and glial cells and macrophages in the optic nerve; a cherry-red spot occurs occasionally. The optic atrophy of Krabbe’s disease (formerly considered one of the familial diffuse scleroses) is due to extensive demyelination associated with the accumulation of globoid bodies containing ceramide-galactose resulting from beta-galactosidase deficiency.
The ceroid lipofuscinoses are among the most commonly inherited childhood neurodegenerative disorders. These autosomal recessive syndromes are caused by accumulation of an insoluble, complex lipopigment that demonstrates autofluorescence and appears microscopically as characteristic curvilinear bodies. Histopathologic examination discloses degeneration of rods and cones beginning at the macula, narrowing of retinal vessels, atrophy of pigment epithelium with pigment migration, and curvilinear bodies in ganglion cells. These progressive disorders are not genetically homogeneous and are separable into infantile, late infantile, juvenile, and Finnish variants. These share seizures, motor disturbances, visual impairment, dementia, and autosomal inheritance.31
The late infantile type (Bielschowsky-Jansky) is a fatal recessively inherited disorder with genomic defect localized to chromosome 13q22, thus delineating this disease as a separate entity.32 Presentation is with developmental arrest and seizures at 2 to 5 years of age, with motor and visual symptoms thereafter. There is massive tissue accumulation of lysosomal hydrophobic subunit-c protein of the mitochondrial adenosine triphosphate synthase. Ganglion cells are decreased in number, with thinning of the nerve fiber layer and optic atrophy. In addition, there is involvement of the outer segments with degeneration of the rods and cones and pigmentary clumping in the outer retinal layers. On MRI, hyperintense periventricular signals correlate with severe loss of myelin.33
Juvenile amaurotic idiocy (Batten-Mayou, Vogt-Spielmeyer) presents as visual loss in early adolescence and is characterized by retinal pigmentary changes predominantly at the macula, with late and minimal pallor of the disc. Apoptosis of photoreceptors and brain neurons has been confirmed as the mechanism of cell death.34 At a later stage, motor and mental deficits evolve. The late or adult form of amaurotic idiocy ascribed to Kufs paradoxically is associated neither with amaurosis nor with retardation to the degree of idiocy, but rather, with dementia with cerebellar and extrapyramidal motor signs.
Optic nerve involvement may occur in the mucopolysaccharidoses (MPS), taking the form of optic atrophy or papilledema. Although true papilledema doubtlessly occurs in association with hydrocephalus seen in these disorders, there are other instances of “papilledema” in which fundus descriptions or photographs are not convincing. Goldberg and Duke35 reported the ocular histopathologic findings in a patient with Hunter’s syndrome (MPS II), in which premortem examination included the observation of “bilateral chronic papilledema.” On microscopic examination, the optic nerve was normal, despite marked retinal pigmentary degeneration, showing neither consecutive atrophy nor changes compatible with chronic papilledema. Similarly, Kenyon and colleagues36 reviewed the systemic mucopolysaccharidoses and included an instance of a 26-year-old man with Hunter’s syndrome (MPS II), whose ophthalmoscopic examination revealed “blurred disc margins in both fundi (without venous congestion, hemorrhages or exudates) compatible with mild chronic papilledema,” but no further comment was made in elucidation of this finding. Hunter’s syndrome is an X-linked recessive disorder characterized by facial and skeletal dysmorphism, stiff joints, and mental slowing. Deafness and chronic disc edema occur, without raised intracranial pressure, and mucopolysaccharide deposition in the sclera and optic nerve septa, especially at the lamina cribrosa, is described.37 MRI may disclose large multifocal cystic areas of hypointense or hyperintense signals in white matter, including the corpus callosum, likely reflecting deposition of mucopolysaccharide and increased fluid content.38
Mailer39 reviewed 16 patients with optic atrophy in gargoylism and concluded that communicating hydrocephalus was the most frequent cause. Although hydrocephalus has long been recognized to occur in mucopolysaccharidosis, it is uncovered more frequently at autopsy than clinically. Goldberg and associates40 reported a case of Maroteaux-Lamy syndrome (MPS VI) with hydrocephalus and papilledema, treated with a ventriculojugular shunt. Those authors discussed the following possible pathogenetic mechanisms of visual dysfunction due to optic nerve involvement in mucopolysaccharidosis: optic atrophy secondary to glaucoma, retinal pigmentary degeneration, or mucopolysaccharide deposition in the retinal elements; infiltration of the nerve substance or meninges; narrowing of the optic canals; and hydrocephalus, with or without papilledema. Retinal pigmentary dystrophy occurs in MPS I-H (Hurler), MPS III (Sanfilippo), and MPS I-S (Scheie), all resulting from storage of heparan sulfate.
The cherry-red spot myoclonus syndrome is an autosomal recessive oligosaccharidosis due to deficiency of lysosomal acetylneuraminidase. The syndrome is comprised of typical cherry spot macula, resting and intention myoclonus, and preserved intellect. Sogg et al41 also described flutterlike ocular oscillations attributed to possible cerebellar involvement.
RETINAL ARTERIAL OCCLUSIONS
Retinal arterial occlusions of neuro-ophthalmologic pertinence occur in patients with carotid athero-occlusive disease, heart disease, arteritis (giant cell, collagen vasculitides), and, rarely, migraine. Cranial arteritis is considered in a subsequent discussion of ischemic optic neuropathies (see Part II of this chapter).
The onset of retinal arterial occlusion rarely goes unnoticed by the patient, but unlike venous thrombosis, arterial occlusions of a minor degree may be difficult to discern ophthalmoscopically, especially if days or weeks pass before the fundus is examined. Muci-Mendoza et al42 used fluorescein fundus angiography to demonstrate ophthalmoscopically occult emboli and post-embolic endothelial damage after episodes of amaurosis fugax (see below, Carotid Atheromatous Disease).
With acute infarction, the retina becomes opaque and takes on a creamy or gray appearance. Atheromatous material in the form of “bright plaques” of cholesterol or other microemboli may be seen, especially lodged at arterial bifurcations (Fig. 5). Segmental arteriolar mural opacification (see Fig. 5D) may follow retinal microembolization by weeks to months, and such sheathing may be as useful as the recognition of the cholesterol embolus itself.43 ERG may show diminished B-wave amplitude, a finding indicating inner retinal ischemia. Weeks following retinal infarction, the optic disc becomes pale, and the arterial tree becomes narrowed in the sector corresponding to the arterial occlusion.
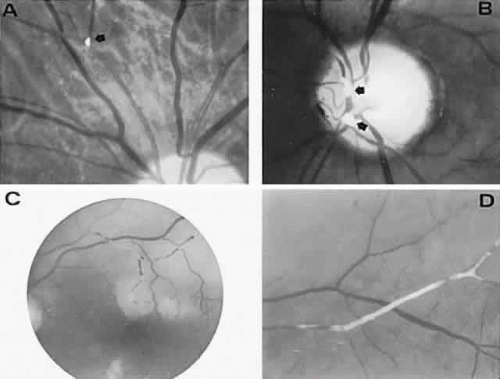 Fig. 5. Retinal microembolic phenomena. A. Bright cholesterol plaque (arrow) impacted at an arterial bifurcation. Thin crystal does not obstruct flow. B. Cholesterol crystals in disc vessels (arrows). Often, the plaque appears larger than the vessel diameter. C. Infarcted opaque retina. The artery contains emboli (? fibrin platelets) that have obstructed flow. D. Reactive opacification of the arterial wall. Fluorescein angiography demonstrated flow through this formerly occluded vessel. |
The visual field defects with arterial occlusion are variable, but they usually take the form of arcuate scotomas or “altitudinal hemianopias” of the superior or inferior half fields (Fig. 6). These altitudinal or pseudo-quadrantic defects are dense and are easily discovered by hand- or finger-counting confrontation techniques. The localizing value of the position of the “vertex” of quadrantic and wedge-shaped defects was pointed out by Alfred Kestenbaum. When the wedge originates at or points toward the blind spot, the defect is due to a retinal arterial occlusion or a lesion at the edge of the optic disc (including the arcuate defects seen in glaucoma). The differential diagnosis of arcuate scotomas, that is, with radial borders originating at the blind spot (see Fig. 6, right field), includes glaucoma, ischemic optic neuropathy, branch retinal artery occlusion, hyaline bodies of optic disc, congenital optic pit, juxtapapillary inflammation, and, rarely, chiasmal interference (see Volume 2, Chapter 6, Fig. 1F).
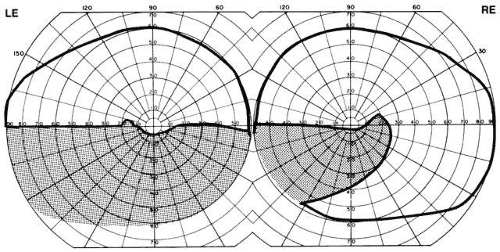 Fig. 6. Field defects of vascular origin. RE. Arcuate nerve fiber bundle defect of the right eye extending from the blind spot into the nasal quadrant. LE. Inferior altitudinal “hemianopia” of the left eye. These patterns are common to both retinal arterial occlusions and segmental infarction of nerve head (ischemic optic neuropathy). |
CAROTID ATHEROMATOUS DISEASE
The pathogenesis of embolic retinal arterial occlusions is diverse, but most are associated with atheromatous degeneration of the cervical carotid arteries. Since the observations by Fisher44 of mobile intravascular material during episodes of transient retinal ischemic episodes (amaurosis fugax), an embolic source is commonly invoked, especially when any material is detected ophthalmoscopically within the retinal circulation. Such retinal emboli have been examined histopathologically and may consist of cholesterol crystals,45 platelet aggregates,46 fibrin and blood cells,47 and neutral fat.48 A study of the prevalence of asymptomatic retinal emboli among 3654 persons aged 49 years or older disclosed photographic evidence in 1.4%, the majority being judged as cholesterol; risk for subsequent stroke is not yet calculated.49 On the other hand, the incidence of visible emboli in retinal arterial occlusion is estimated at about 20%.50 Wilson and coworkers51 compared the incidence of carotid and cardiac disease in 103 patients with retinal artery occlusions and reported cardiac valve abnormalities in 28; of 62 carotid arteriograms, 35% were normal, 13% were occluded, and the remainder showed irregularity or significant stenosis. In a small group of 41 patients with transient monocular visual loss or retinal artery occlusion, no cardiac or carotid source was uncovered in two-thirds of subjects, ipsilateral carotid disease was seen in 11 (27%) cases, and a cardiac source was noted in a single patient.52
Even in those patients without ophthalmoscopically visible emboli, it is possible that the embolus either has lodged in the retro-laminar portion of the optic disc or has disintegrated and passed to the retinal periphery. Primary thrombosis in the retinal arterial circulation, beyond evidence of frank vasculitis or hypercoagulable states, is considered obsolete. Vasospasm, on the other hand, is an unlikely candidate to explain arterial occlusion, although rare instances of otherwise unexplained transient visual loss are reported.53 Other noncarotid sources of emboli, including cardiogenic, are discussed below.
Transient ischemic episodes involving the retinal arterial tree produce the well-known symptom of amaurosis fugax (“fleeting darkness, or blindness”; ocular transient ischemic episode). This phenomenon may be defined as a painless unilateral, transient loss of vision that usually progresses from the periphery toward the center of the field. Often, the visual deficit takes the pattern of a dark curtain descending from above or ascending from below. Complete or subtotal blindness follows in seconds and lasts from 1 to 5 minutes (rarely longer). Vision returns to normal within 10 to 20 minutes, at times by reversal of the pattern of progression. Incomplete variations produce less distinctive sensations such as “looking through a fog … through raindrops … through haze.” By convention, the term amaurosis fugax is reserved for the ocular symptoms as described, which are usually distinguishable from other episodic visual disturbances (see Volume 2, Chapter 1), and it implies transient ischemia in retinal arterial circulation. Those conditions with principally monocular transient visual loss are listed in Table 3.54
It is likely that microembolic material in the form of fibrin-platelet aggregates momentarily occludes retinal vessels, then fragments and passes into the retinal periphery. If disaggregation with reconstitution of blood flow does not take place within several minutes, ischemic damage to inner retinal layers may be irreversible, and permanent visual defects may ensue. Most evidence indicates that the major source of retinal microemboli is the extracranial internal carotid artery, specifically an ulcerating atheromatous lesion at the level of the bifurcation.55 The incidence of carotid atheromatous disease in patients with retinal strokes or amaurosis fugax, as noted above, ranges from a low of 27%52 to an estimated high from 57 to 67%.56 Moreover, retinal ischemic events may be more frequent when carotid stenosis is greater than 50% to 70% or in the presence of ulcerative atheromatous plaques.57,58 The diagnosis of amaurosis fugax and the implied underlying carotid atheromatous disease may usually be made on the basis of signs and symptoms (Table 4).
TABLE 4. Symptoms and Signs of Carotid Atheromatous Disease | ||||||
|---|---|---|---|---|---|---|
|
The presence of a carotid bruit is suggestive evidence of turbulent flow, but, however practical, auscultation and tender palpation of the carotid arteries and performance of ophthalmodynamometry or ocular plethysmography no longer suffice. At any rate, in the patient with amaurosis fugax, in whom carotid disease must be evaluated, further technical procedures are mandatory. Such tests of carotid morphology and hemodynamics assume two distinct forms: arteriographic, requiring intraluminal contrast opacification of arteries; and nonarteriographic (“noninvasive”), using ultrasound, Doppler imaging, and, most recently, MRI (magnetic resonance angiography or MRA). Noninvasive panels are commonly employed for preliminary assessment, with the advantages of risk-free, outpatient application. Carotid duplex scanning combines simultaneous real-time B-mode ultrasonography with gated, pulsed Doppler ultrasonography. These studies provide color-coded images of the degree of stenosis and plaque morphology, although not without limitations and artifactual data.59 MRA imaging of carotid vessels is currently an evolving field, but preliminary drawbacks include overestimation of stenosis and perturbation by signal voids related to turbulant flow. MRA images are useful in combination with duplex ultrasonography, especially when the carotid bifurcation is involved.58
The general consensus58 is that there is a sharp decline in the risk of stroke and in benefit from endarterectomy as the degree of angiographically defined stenosis diminishes from 99% to 70%, and this critical point underscores the importance of precise measurement of stenosis. Whereas modern techniques of angiography carry a 0.09% to 0.3% stroke rate risk, it is generally recognized that duplex ultrasonography be used as a screening tool to exclude patients with no detectable carotid disease from further testing, but patients suspected of harboring carotid disease, who are otherwise suitable candidates for endarterectomy, should undergo confirmatory contrast angiography. Ultrasonography may also be applied to monitor for progression of stenosis. Therefore, the elimination of conventional carotid angiography in favor of combined Doppler and MRA, to assess for stroke risk or determination of proper therapy, is premature, cost-containment strategies notwithstanding.
In contrast to transient symptoms and signs of retinal microembolic episodes, a condition of chronic ocular hypoxia (ocular ischemic syndrome) occurs less frequently, resulting from diffuse vascular occlusive disease of the aortic arch or common carotid artery. Acute or chronic occlusion with insufficient collateralization produces an ischemic pseudo-inflammatory uveitis, which variably includes an injected painful globe, corneal edema, aqueous flare and cells, a mid-dilated fixed pupil, rubeosis and iris atrophy, rapidly advancing cataract, either hypotony or elevated intraocular pressure (“neovascular glaucoma”), retinal microaneurysms and new vessel formation, posterior pole and mid-peripheral blot hemorrhages, macular edema, venous dilation and “sausaging,” cytoid infarcts (cotton-wool spots) of the nerve fiber layer, and arterial occlusions (Fig. 7; see Table 4). The hypoxemic fundus changes constitute a picture of venous stasis (low-pressure) retinopathy, perhaps the commonest ocular sign of chronic carotid obstruction.
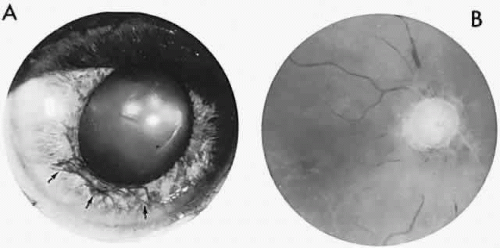 Fig. 7. Ocular hypoxia with subacute carotid occlusion. The patient complained of a painful red eye. A. Anterior segment shows an irregular, fixed pupil and iris rubeosis (arrows). B. Fundus demonstrates combined retinochoroidal infarction with acute excavation of the optic disc. Arteriography revealed right internal carotid occlusion. |
Ischemic photoreceptor metabolism accounts for subjective afterimages following exposure to bright light, including a positive photostress test60 (see Volume 2, Chapter 2). Low retinal arterial pressure may be detected by observing pulsation or collapse of the disc arterioles with even slight fingertip pressure exerted on the globe. Such borderline perfusion associated with carotid stenosis and retinopathy may be heralded by postprandial visual loss.61 In the situation of chronic, subacute, or rapidly progressive ischemic oculopathy, giant cell arteritis must be considered in the differential diagnosis, and chronic venous obstruction or diabetic retinopathy may produce similar fundus appearance.
From a series of 32 patients62 with ocular ischemic syndrome manifested by anterior segment neovascularization, the following data were accrued: mean age 68 ± 8 years; visual symptoms presented as amaurosis fugax, 15%, gradual, 28%, or sudden, 41% loss; initial visual acuity less than or equal to 20/400 in 64%, and in 77% at final follow-up; iris neovascularization, 87%; iridocorneal angle neovascularization, 59%; disc pallor, 40%, or cupped, 19%, or edematous, 8%; retinal circulatory stasis, 21%; retinal hemorrhages, 24%. Intraocular pressures ranged widely from 4 mmHg to 60 mmHg. Ipsilateral carotid occlusion or severe stenosis (80% to 99%) was found in 74%, but endarterectomy in 7 patients did not influence visual outcome, which was poor at onset. Other significantly associated systemic diseases included diabetes (56%), arterial hypertension (50%), coronary artery disease (38%), and previous stroke or transient ischemic attack (TIA) (30%).
Visual outcome in such ischemic globes is guarded, but endarterectomy may prevent progressive infarction of ocular tissues and may alleviate pain (ocular “angina”). Improvement or stabilization of vision and some resolution of retinopathy are reported, especially if carotid reconstruction is performed before irreversible neovascular glaucoma.63 A vasospastic form of ocular ischemia is reported, with improvement with the calcium channel blocker verapamil.64
It is exceedingly difficult to predict which patients with amaurosis fugax will develop permanent visual loss or suffer a hemispheral vascular accident, and it is not yet clear that patients with asymptomatic carotid stenosis benefit from arterial surgery. From the exacting North American Symptomatic Carotid Endarterectomy Trial (NASCET),65 in patients with 70% or greater stenosis and cerebral TIAs within the past 4 months, stroke risk is estimated at up to 25% per year; with first-time amaurosis fugax and high-grade stenosis, the risk is 8.5% per year, but apparently with less severe stoke deficits.66 These risks are compared with surgical complication rate of carotid endarterectomy, with a perioperative stroke or death rate of 5.8% in the NASCET study, but as much as 8.5% even in academic centers,67 and the surgical morbidity/mortality figures may be greater outside major vascular surgery facilities.
Regarding the Asymptomatic Carotid Atherosclerosis Study (ACAS),68 in 1662 patients with greater than 60% stenosis but no symptoms, after 4465 cumulative patient-years, aggregate risk of stroke or death was 4.8% among surgical patients and 10.6% among patients who did not have surgery; both groups received aspirin and attempts at reduction of risk factors; perioperative morbidity/mortality risk was 3%, but patients were selected to avoid those with confounding factors to avoid excessive surgical risk. Interestingly, patients with 80% stenosis or greater had a lower subsequent events rate than those with less stenosis.
To reiterate, carotid endarterectomy may be recommended for properly selected asymptomatic patients with stenosis of 80% to 90% by modern contrast angiography and when angiography and surgery can be performed with a combined stroke or death rate of less than 3%. In patients with asymptomatic carotid stenosis who are prepared to undergo coronary artery bypass grafting for symptomatic coronary artery disease, there are no data suggesting benefit from prophylactic endarterectomy.69
In a British study,70 retinal infarction was believed to have some prognostic value, the presence of retinal emboli being associated with increased mortality rate of 8% per year. It was emphasized that the most deaths in such patients were related to cardiac infarctions.
In recent years, aspirin, in daily low doses of 75 mg to 325 mg, has been used as a platelet antiaggregant. A collaborative overview71 summarizing data from 173 randomized trials of antiplatelet therapy in patients at high risk for occlusive vascular disease showed a definite protective value for myocardial infarction, nonfatal stroke, and vascular death, with respective risk reductions of one-third, one-third, and one-sixth. Other previous analyses showed stroke reduction rates as high as 42% in both men and women; dipyridamole (Persantine) has no established role in stroke prophylaxis.72 Most interestingly, an analysis of published randomized studies of the medical treatment (chronic anticoagulation or platelet inhibitors, versus no treatment or placebo) of TIAs showed that neither treatment modality significantly reduced mortality rates.73 Failure to prevent cardiac complications appears independent of therapeutic effectiveness in reducing the incidence of cerebral strokes in patients with TIAs.
Not without controversy, there is general agreement with the following management recommendations regarding indications for endarterectomy, as modified from Trobe.74 Patients for whom carotid endarterectomy is indicated: cerebral hemispheric ischemic symptoms within the carotid artery distribution occurring within the past 6 months; ipsilateral stenosis of 70% or greater, without intraluminal thrombosis or substantial syphon stenosis (i.e., distal intracranial segment stenosis does not exceed cervical segment stenosis); age less than 75 years; no evidence of significant disease (organ failure) of kidney, liver, lung; no severe disabling stroke or progressing neurologic dysfunction; no recent myocardial infarct, unstable angina pectoris, or a cardiac valvular or rhythm disorder that could be a source of embolic symptoms; no uncontrolled hypertension; life expectancy of at least 2 years and adequate quality of life. Patients with acute ocular ischemia (amaurosis fugax, recent retinal infarction, ischemic optic neuropathy) do not qualify. Again, it is imperative that a proficient vascular surgical team with a perioperative stroke and death rate less than 2% to 4% should be available. It also must be reiterarated that atherosclerotic disease is multifocal, and most patients succumb to myocardial infarcts, not stroke.
Other nonatheromatous carotid diseases may manifest as transient visual obscurations. Fibromuscular dysplasia, most common in younger women, is also a likely source of recurrent microembolization, requiring angiography for confirmation, and surgical intraluminal dilation may be required.75 Spontaneous dissection of the cervical segment of the internal carotid artery is increasingly recognized as a cause of stroke, with a mean age of 45 years (range, 16 to 76 years), and an estimated annual incidence of 2.5 to 3.5 per 100,000.76 Symptoms of dissection frequently include headache, neck and jaw pain, dysphagia, metallic dysgeusia, Horner’s syndrome, and amaurosis fugax. According to Biousse and colleagues,76a nearly two-thirds of patients have ocular signs or symptoms, 5,270 at presentation; almost half have a painful Horner’s syndrome, one-third with transient monocular vision loss, and rarely ischemic optic neuropathy occurs. Trivial trauma or exertion such as weight-lifting may be inapparent contributing factors, but predisposing conditions include fibromuscular dysplasia, Marfan’s or Ehler-Danlos syndrome, and cystic medial necrosis.77 Diagnosis is made on angiography, which shows irregular narrowing, and MRI discloses hyperintense signal of mural hematoma. Duplex echography demonstrates arterial enlargement due to mural hematoma and stenosis distal to the point of dissection. Recanalization is the anticipated outcome, but antithrombotic therapy such as heparin is usually used to prevent subsequent embolization.
Pulseless disease (Takayasu’s arteritis), suggested by the inability to obtain peripheral pulses in the arms and confirmed by aortic arch angiography,78 is also associated with transient obscurations of vision and chronic retinal ischemic changes. This disorder is an idiopathic chronic inflammation of the aorta and proximal segments of its major branches, producing progressive stenosis and end-organ hypoperfusion. Although there is a predilection for Asians and for women, pulseless disease has been described in all racial groups. Ophthalmologic signs and symptoms are considered late manifestations, and visual loss may be noted when the patient assumes an erect posture. Ischemic retinopathy, iris neovascularization, cataract, vitreous hemorrhage, and anterior segment ischemia are chief ophthalmic findings.
Color Doppler ultrasound imaging is a relatively recent noninvasive technique to assess blood flow dynamics in the eye and orbit, although the accuracy and applicabilities are not yet fully explored. Nonetheless, preliminary investigations suggest effective application in retinal arterial occlusions, cranial arteritis, ischemic neuropathies, and carotid artery disease.79 In a study of 24 persons with greater than 75% carotid stenosis,80 all patients showed lower mean peak systolic velocities in the central retinal, posterior ciliary, and ophthalmic arteries, with improvement after endarterectomy.
OTHER RETINAL ARTERIAL OCCLUSIONS
Although atherosclerotic disease of the extracranial carotid system is by far the most common source of emboli to the retina and brain, other sites should be considered. Embolic material may originate from damaged endocardium following myocardial infarction, a significant difference being noted between observed and expected probabilities of stroke at 1 and 2 months.81 Rheumatic or atherosclerotic aortic and mitral valvular disease may serve as a nidus for recurrent embolus formation. Patent foramen ovale is found in one-third of normal hearts at autopsy, as well as other septal defects (right-to-left shunts), and atrial fibrillation, are all potential sources of retinal and cerebral microemboli. In recent years, mucoid degeneration of the mitral valve and chordae tendineae (“prolapsing mitral valve”; Barlow’s syndrome) has been recognized as a source for ocular and cerebral stroke and transient ischemic events.82,83 In 59 patients with mitral prolapse, Lesser and colleagues83 found an incidence of 22% with amaurosis fugax. This syndrome should come to mind especially in nonhypertensive patients younger than the sixth decade; both men and women may be affected, and most patients with native valve endocarditis have mitral valve prolapse.84 This condition is suggested by chest pain, dyspnea and cardiac arrhythmias, and mid-systolic click or murmur. The diagnosis is confirmed by echocardiography and angiocardiography. Otherwise, endocarditis of native or prosthetic valves is rarely unaccompanied by systemic manifestations of malaise, fever, petechiae, and heart murmur. Other ocular manifestations include conjunctival petechiae, Roth’s spots, choroidal septic emboli with subretinal vascularization, embolic retinitis, and endophthalmitis.85
Retinal arterial obstructions in children and young adults are only rarely the result of embolization in the absence of detectable cardiac disease. Of 27 patients with retinal artery occlusions occurring before the age of 30 years, Brown and colleagues86 found a history of migraine headaches in 8 patients, but no instance of a previous history of “retinal migraine” attacks (see below), and emboli were detected in only 7%. In a similar series of retinal arterial occlusions in 27 eyes of 21 persons aged 22 to 33 years, 67% of whom were women, Greven and coworkers87 found identifiable emboli in 7 (33%) patients and cardiac valvular disease (atrial myxoma, bacterial endocarditis, mitral valve lesions) in only 4; hypercoagulable or embolic factors included various admixtures of oral contraceptives, cigarette use, pregnancy, obesity, and migraine; 2 patients with either anticardiolipin antibody elevation or protein S deficiency were both pregnant, and no patient in the series had typical migrainous symptoms at the time of retinal arterial occlusion. The role of transesophageal echocardiography is stressed, especially in young patients with unaccountable arterial occlusions, with or without visible emboli.88 In a retrospective review89 of 16 patients with idiopathic recurrent branch arterial occlusions and no particular common risk factor, the long-term visual, neurologic, and systemic prognosis remained favorable after a mean follow-up of 9 years, with no systemic thromboembolic events.
Coagulation studies (e.g., prothrombin and partial thromboplastin times, total platelet count) are generally unrevealing unless specific thrombophilic (coagulants and fibrinolytics) factors are evaluated, such as lupus anticoagulants, other immunoglobulins against phospholipids, proteins C and S (inhibit clotting cascade), antithrombin III, and homocysteine.90,91 The antiphospholipid antibody syndrome accounts for both venous and arterial occlusions. Antibodies to negatively charged phospholipid include anticardiolipin antibodies, lupus anticoagulant, and those responsible for biologic false-positive Venereal Disease Research Laboratory tests. Antiphospholipid antibodies are detectable in 2% of healthy persons, but in 35% to 50% of patients with lupus erythematosus and in 25% to 50% of young patients with stroke.92 However, in a prospective study93 of 75 patients with retinal vascular occlusions, mostly venous, no increased prevalence of antiphospholipid antibodies was found. With regard to homocysteine, there is unequivocal evidence that hyperhomocysteinemia is a risk factor in carotid artery stenosis,94 likely related to impaired production of endothelium-derived relaxing factor, to stimulated proliferation of smooth muscle cells that play a key role in atherogenesis and affecting the expression of thrombomodulin and activation of protein C.
Among young patients with stroke, 17% exhibit a deficiency of natural anticoagulants, with protein S deficiency in 12%, protein C deficiency in 2%, and antithrombin III deficiency in 2%. However, 5 to 10 times more common than these conditions is functional resistance to activated protein C (APC), especially in youthful patients with venous thrombosis, and it is prevalent in the general population in 2% to 5% of individuals. APC, also known as factor V Leiden, is due to a single point mutation altering coding for residue 506 from arginine to glycine and is dominantly inherited.92
Branch arterial occlusion is documented in association with Lyme disease,95 and arterial and venous retinal vascular disease is rarely associated with Crohn’s ulcerative colitis.96 Other systemic (“collagen”) vasculitides such as lupus erythematosus, polyarteritis nodosa,97 and dermatomyositis are also infrequent causes of retinal arterial occlusions, but they must be suspected especially in young women. Serum complement (C3, C4) and antinuclear antibody (ANA) levels, erythrocyte sedimentation rate (ESR), and other rheumatologic evaluations are mandatory.
The single and multiple influences of some risk factors remain speculative. For example, the previously described mitral valve prolapse syndrome, with or without “sticky platelets,” occurs in some 20% of otherwise healthy women, migraine affects at least 10% (other estimates run as high a prevalence as 20% in men and 30% in women) of the population, and millions of women regularly use oral anovulatory agents.
It bears emphasizing that the origin of transient, monotonously stereotyped visual obscurations in healthy young persons often proves elusive, but fortunately most of these attacks are self-limited and benign, with neither identifiable risk factors nor need for therapy. Of course, echocardiography should be considered. Extrapolating from general stroke data,98 there is no consensus that low-dose oral contraceptives increase the risk of retinal vascular occlusions. However, other reviews99 suggest that the presence of complex or prolonged migraine aura, or of additional stroke risk factors (increased age, smoking, hypertension), likely increases the ischemic stroke risk further in patients with migraine when oral contraceptives are added. Otherwise, intra-ocular pressure, occult vasculitis, and hemoglobinopathies should be considered in patients with retinal arterial occlusions, as well as the multiple causes considered here, and judiciously selective laboratory assessments should be applied. That is not to say that definitive diagnosis is forthcoming, nor do abnormal laboratory data necessarily imply cause and effect; for example, antiphospholipid antibodies are found in healthy persons. Optimal, or even minimal, therapy remains controversial, and only basic regimens currently suffice: systemic corticosteroids to suppress antibody production; anticoagulants to block thrombosis; and antiplatelet agents. In the acute period, perhaps hours after occlusion, ocular massage may lower intraocular tension, and microcatheter infusion of urokinase or tissue plasminogen activator is advocated,100 but there is little evidence to support anterior chamber paracentesis or other medical therapies.85
Retinal vein occlusion has far fewer neuro-ophthalmologic implications than do arterial retinal infarctions. Significant associations are found with hypertension, diabetes, glaucoma, cardiovascular disease, and increased ESR in women, but not with estrogen use, alcohol consumption, or physical activity.101 Vein occlusion is reported in association with hyperhomocysteinemia,102 and with antiphospholipid antibody syndrome,103 although in larger series94 no direct relationship with anti-cardiolipin antibodies or lupus anticoagulant is found. However, in patients younger than 45 years, dominantly inherited APC resistance (see above) is a distinct risk factor.104 Venous stasis retinopathy may be found in instances of arteriovenous shunts or fistulas, as discussed in Volume 2, Chapter 17. The general problem of venous occlusions is reviewed elsewhere.105
RETINAL MIGRAINE
Retinal migraine implies stereotypical transient monocular loss of vision of rapid onset, usually followed by ipsilateral headache, in persons usually with other migrainous symptoms, including typical common migraine headaches. The following case history is exemplary:
A 22 year-old male medical student complained of “blackouts” in the left eye, lasting between 10 and 20 minutes, often followed by mild diffuse headache localized to the left side of the head. Cephalgia usually began as the visual deficit was clearing. These episodes began at age 16, at first occurring once or twice per year, but currently every 4 weeks for the past 6 months, especially at “exam time.” The patient had no other particular headache pattern, and the family history was negative. Standard cardiac echography was unremarkable.
The retinal variety may be admixed in a person who suffers the more conventional attacks of migraine. It is presumed that vasospasm in the retinal circulation determines transient hypoxia, perhaps somewhat similar to the visual cortical event. On rare occasions, the fundus has been examined during typical retinal migraine episodes, and arterial constriction has been described. Wolter and Burchfield106 photographically documented such an episode and demonstrated mild “retinal edema”; vessel narrowing is also evident (Fig. 8). Fortunately, permanent complications of retinal migraine are rare. These may take the form of central retinal artery occlusion or ischemic papillopathy (see Volume 2, Chapter 16); nerve fiber bundle visual field defects may be demonstrated (Fig. 9).
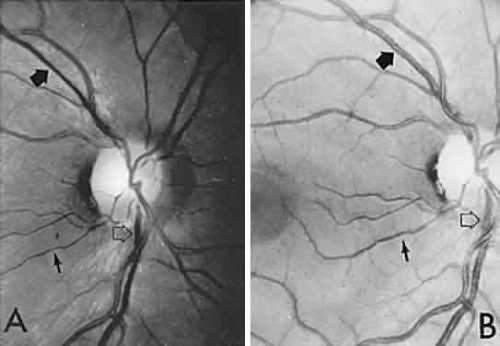 Fig. 8. Retinal migraine. A. During amaurotic episode. Note the dusky appearance of the fundus, increased retinal sheen (possibly edema), and dark narrowed veins (arrows). The disc is also hyperemic. B. Fundus after episode. Compare paired arrows. (Courtesy of Dr. J. Reimer Wolter) |
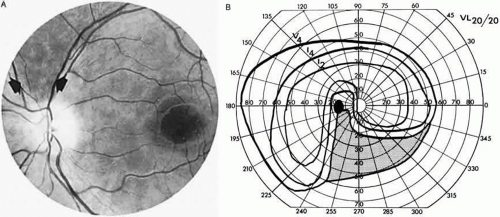 Fig. 9. An 18-year-old student with recurrent episodes of left retinal migraine. After a typical attack, he noted an inferior field defect. A. Fundus shows a defect in the superior arcuate nerve fiber bundle (between arrows: compare fiber layer below disc). B. Visual field defect corresponds to a retinal nerve fiber layer defect. |
RETINAL VASCULITIS
Mild monocular visual blurring and a fundus picture of disc swelling with dilated veins and multiple small nerve fiber layer hemorrhages constitute a problem in diagnosis and management. In young patients whose vision is retained at good levels and the abnormality spontaneously regresses, a diagnosis of central retinal vein occlusion (see above) is usually made, without disclosure of underlying inflammation, hyperviscosity, or coagulopathy.
When disc swelling seems disproportionate to other hemorrhagic features, the vague term “papillophlebitis” is clinically applied, admittedly with little or no evidence of actual inflammatory disease. Although the course may be protracted (up to 18 months), the outcome is benign and apparently is unaltered by corticosteroid therapy. In a series of 40 patients with this diagnostic rubric,107 some 7 cases of mitral valve prolapse were found, and 8 of 23 patients studied hematologically showed an increase in platelet coagulant activity concerned with the initiation of early stages of intrinsic coagulation. Nine patients had macular edema that contributed to lowered acuity, at times worse than 20/200, and “all patients had a fluorescein angiogram that was consistent with a central retinal vein occlusion of the nonischemic type.” So-called papillophlebitis may be associated with retinal arterial occlusions and may possibly represent a form of idiopathic vasculitis or a coagulation defect of pregnancy.108
In contrast to this benign form of papillary or retinal “vasculitis,” Cogan109 documented a “severe vasculitis” category, including cases of periarteritis nodosa, Behçet’s disease, multiple sclerosis, and granulomatous vasculitis. The cause in patients without systemic disease is speculative, but Cogan demonstrated in his patients with “severe vasculitis” that there was round cell infiltration of the venular walls.
The question of retinal arteritis, vasculitis, and autoimmune mechanisms was admirably reviewed by M. D. Sanders,110 who presented clinical information on 150 cases examined at St. Thomas’ Hospital, London. Retinal vasculitis should be considered when inflammatory changes (focal or extensive sheathing or occlusion of vessels; retinal infiltrates and hemorrhages; cellular debris in vitreous; vascular leakage of fluorescein) occur in relation to retinal vessels. Diagnostic categories include the following: infectious (tuberculosis, syphilis, herpes simplex, cytomegalovirus [CMV]); multiple sclerosis with phlebitis; polyarteritis nodosa, lupus erythematosus, Wegener’s and Goodpasture’s syndromes, sarcoidosis, Behçet’s disease; autoimmune vasculitis without systemic disease, polymyositis, dermatomyositis, polyarteritis nodosa, Whipple’s disease, and ulcerative colitis. Despite these purported associations, in well patients with primary retinal vasculitis and no medical history suggestive of underlying systemic disease, results of diagnostic batteries are meager, and follow-up data do not suggest subsequent manifestations of specific causes.111 Although it is admittedly unrewarding, minimal evaluation would include complete blood count, ESR, urinalysis, fluorescent treponemal antibody absorption test, rapid plasma reagent, and chest roentgenogram.111
Retinal vasculitis exceptionally is associated with CNS vasculitis (angiitis). In 1882, Eales described a variety of retinal periphlebitis characterized by “retinal hemorrhage associated with epistaxis and constipation” seen in young men in southern England. Patients present with recurrent, usually monocular, vitreous hemorrhages that may persist for years and ultimately involve the second eye. Neurologic complications must be rare, but subacute myelopathies, chronic lymphocytic meningitis, and middle cerebral artery stroke have been reported.112 Distinction from the retinal vasculitides discussed above is problematic, and “Eales’ disease” remains a diagnosis of exclusion.
Microangiopathy of the brain, retina, and inner ear (Susac’s syndrome) is a rare disorder predominantly affecting women of child-bearing age, but without a specific origin or systemic manifestations. An immune or coagulopathic background is unproved. Patients present with the following: vision loss due to branch retinal arteriolar occlusions with vessel hyperfluorescence on fluorescein angiography, and delayed leakage; hearing loss; multiple CNS infarctions.113 Efficacy of treatment with corticosteroids and immunosuppressive agents is uncertain, but hyperbaric oxygenation has been beneficial in a single case, with rapid visual improvement.114
Other infrequent causes of multifocal segmental retinal vasculitis, co-mingled with neuroretinitis and choroiditis, include Lyme disease,115 Rochalimaea infection (cat-scratch disease),116 and intraocular lymphoma.117 Without neurologic or systemic implications, a syndrome of retinal vasculitis, with aneurysmal dilatation of arterioles, capillary nonperfusion, and exudative neuroretinitis with marked decrease in acuity, has been described, with an age range of 9 to 49 years and a female preponderance; oral corticosteroids had no beneficial effects.118 Ten patients with isolated retinal vasculitis with family history of multiple sclerosis, or positive HLA-B7 typing, underwent MRI, which showed white matter lesions resembling those of multiple sclerosis in 3 instances, suggesting a causative relationship.119
UVEO-MENINGEAL SYNDROMES
As noted, surprising numbers of etiologically diverse diseases simultaneously or sequentially involve the retina, uvea, and brain. Of these, the Vogt-Koyanagi-Harada syndrome is best known, characterized by bilateral diffuse granulomatous panuveitis, whitening (poliosis) especially of eyebrows and lashes, skin depigmentation (vitiligo), alopecia, meningismus with headache and CSF pleocytosis, rarely focal CNS signs, tinnitus, hearing loss, and vertigo. Autoimmune inflammation directed against melanocytes seems the basic mechanism, with an epidemiologic predilection for pigmented racial groups, especially in Japan and other parts of Asia; it is uncommon in whites. Prolonged corticosteroid and other immunosuppressive therapy is effective.120
Ureitis, including periphlebitis (“venous sheathing”), and pars planitis, is infrequently discovered in association with multiple sclerosis. Delay between onset of neurologic and ocular symptoms occurs, and the possibility of a shared genetic factor is raised.120a
Zonal outer retinopathies (AZOOR, see above) can show choroidal changes and are reported with CSF pleocytosis, cervical myelopathy, and periventricular white matter lesions on MRI, abnormal ERG, perivenous sheathing, and retinal pigment migration.121 Posterior placoid pigment epitheliopathy (APMPPE, see above) also is reported to occur in association with headaches, CSF aseptic cellular reaction, TIAs, inner ear symptoms, and multiple strokes, requiring immunosuppressive agents for presumed cerebral vasculitis.122 The classification found in Table 5 highlights the difficult diagnostic dilemma posed by these diverse disorders.
TABLE 5. Uveo-meningeal Syndromes | ||||||||
|---|---|---|---|---|---|---|---|---|
|
HUMAN IMMUNODEFICIENCY VIRUS INFECTION AND AIDS
The spectrum of ocular, orbital, and CNS involvement with human immunodeficiency virus (HIV) infection, and subsequent acquired immune deficiency syndrome (AIDS), is vast and complicated, characterized by peculiar neoplasia and a host of opportunistic infectious agents that invade the retina, optic nerve, leptomeninges, and brain, at times mimicking other neurologic syndromes. Co-existing manifestations in the eye and brain present confounding factors for neuro-ophthalmologic localization. The usual parsimonious medical expectation that a single basic disorder encompasses all manifestations of an illness is inoperative in the patient with severely depressed cellular immunity, and, indeed, unrelated problems may be mislabeled. Ocular findings are detected in the majority of patients with AIDS at sometime in the course of the disease, the most common being relatively asymptomatic noninfectious microangiopathy consisting principally of microinfarcts (“cotton-wool spots”) admixed with small hemorrhages, occurring in at least 50% of patients with AIDS, 34% of those with AIDS-related complex, and 3% of persons with asymptomatic HIV infection.123 Interestingly, there is distinct evidence124 of abnormal visual function (contrast sensitivity function, automated perimetry, color sense) in HIV-positive patients without ophthalmoscopic evidence of retinopathy, with normal global neuropsychologic function, and unrelated to disease state as determined by markers including CD4 T-lymphocyte count. It is speculated that such visual function may be related to HIV infection at some level of the visual pathways, or it may be an effect of antiviral or other therapeutic agents. Morphometric techniques125 have demonstrated markedly lower mean axonal populations in AIDS-affected optic nerves, possibly reflecting a form of primary optic neuropathy (see Part II of this chapter for discussion of optic neuropathies in AIDS).
Opportunistic infections, particularly CMV retinitis, are major causes of severe visual loss and blindness; CMV retinitis is estimated to occur in 37% of patients with AIDS.126 CMV retinitis is a relatively late manifestation of the basic disease and is associated with CD4 T-cell counts of less than 0.10 × 109/L. Other tissues are affected, including the brain, lungs, and gastrointestinal tract. CMV retinitis presents as patches of opaque retina with intraretinal hemorrhages and exudative borders of advancing necrosis, which may eventuate in retinal detachment. Intravenous or intraocular ganciclovir and intravenous foscarnet are effective in controlling (viro-static) CMV retinitis, but they do not eliminate the virus.126
Other opportunistic fundus infections include the following: toxoplasmosis (retinochoroiditis, at times also involving optic nerve127), frequently accompanied by toxoplasmic encephalitis, the most common cause of focal CNS dysfunction in AIDS; varicella-zoster virus, producing acute retinal necrosis128; herpes simplex; and Pneumocystis carinii choroidopathy. Central retinal vein occlusions are documented, with pathologic examination disclosing no histologic evidence of HIV, a finding suggesting other hemorheologic factors.129 The protean manifestations of AIDS are exemplified by a case of sudden blindness in a 50-year-old man who showed, at autopsy, simultaneous CMV infection of the retina, herpes simplex in the optic nerve, and nonHodgkin’s lymphoma of the optic tract.130
TOXIC RETINOPATHIES
Given the vast array of pharmaceutical agents and their extensive usage, and ingenious “recreational” drug usage, toxic effects on the retina only infrequently are encountered (see Part II of this chapter for Toxic Optic Neuropathies). Aside from the well-known problems with observable pigmentary maculopathies secondary to long-term intake of the antipsychotic phenothiazines, and hydroxychloroquine131 (for rheumatoid arthritis, lupus erythematosus), and the acute systemic response to methanol poisoning, included here is retinal toxicity with peculiar symptoms or a clinical course that could be misconstrued in neuro-ophthalmologic context.
Ocular symptoms of the cardiac glycosides have been recognized since the time of Withering, who wrote on “foxglove” in 1785. Visual anomalies include blurring, peri-central scotoma, abnormal dark adaptation, xanthopsia (“yellow vision”), a peculiar sensation of whitish glare described at times as “frosting,” and other aberrations of color perception. Symptoms may be continuous or intermittent and are reversible with diminished dosage levels, although other systemic signs of digitalis toxicity may be absent, and, indeed, serum levels may be well within the normal therapeutic range.132 Although this condition is classically attributed to optic nerve dysfunction, evidence provided by ERG and color vision data implicate a cone dysfunction syndrome, likely related to inhibition of adenosine triphosphate in rod outer segments, or ganglion cell intoxication.132 Pain on eye movement is also reported.133 Fisher134 recorded visual disturbances in five elderly patients who were receiving quinidine therapy, disturbances that were at first attributed to transient ischemic episodes, consisting of temporary dark shadows or bright afterimages noted only on awakening, and lasting a few minutes to less than 1 hour. Four of these patients were also receiving digoxin, and a synergistic effect cannot be dismissed. Although the long-term effect of quinine on retinal ganglion cells is well known, causing severe visual field contraction, optic atrophy, and narrowing of retinal arteries, Fisher considered these morning scotomas to be symptoms of transient failure of retinal light adaptation.
The oral antiestrogen agent tamoxifen, used for breast cancer, even at high daily doses has infrequent ocular side effects, including the deposition of fine crystalline deposits in the retina and occasional macular edema,135 and intra-arterial cisplatin for glioblastoma may produce severe retinotoxic effects.136 Interferon produces retinopathy characterized by hemorrhages and cotton-wool spots.137 In patients undergoing renal transplantation, to prevent organ rejection, a murine monoclonal antibody, OKT3, is used and is associated with profound and irreversible visual loss at the retinal level.138
Most dramatically, temporary blindness occurs following transuretheral prostate resection (TURP), during which a nonelectrolyte, nonconducting irrigating fluid (glycine) is absorbed through prostatic venous sinuses into the systemic circulation.139 The TURP syndrome consists of confusion, bradycardia, nausea, hypertension, convulsions, and visual disturbance (even to no light perception) lasting minutes to several hours, and it may be related to hyponatremia, glycine retinal toxicity, or cortical edema. Retinol (vitamin A) is fat soluble, absorbed in the small intestine, and stored in the liver; deficiency occurs in malnutritional situations, liver dysfunction, and malabsorption states,140 including mastocytosis,141 and it is characterized by night blindness, visual loss, and abnormal ERG findings.
Along with other remote effects of carcinoma on the nervous system must be included an immune-mediated retinal photoreceptor degeneration, cancer-associated retinopathy (CAR). Described initially, and most commonly, with small cell carcinoma of the lung, visual disturbances include usually subacute loss of acuity and field depression, color anomalies, narrowed arterioles, pigment epithelial disturbances, and vitreous cells. The ERG is severely diminished, and pathologic examination discloses loss of rods and cones and thinning of the outer nuclear layer of the retina, but only mild disruption of inner retinal layers. Antiretina antibodies have been isolated in sera from patients with CAR syndrome, and rising titers of CAR antigen prove useful in identifying this disorder.142 Acute night blindness and sensations of shimmering lights have been reported as paraneoplastic effects of melanoma (MAR), with ERG and other psychophysical responses consistent with interruption of bipolar rod function and selective disruption of magnocelluler neurons.143 Thirkill144 provided a review of the CAR syndrome and noted the evidence of elaboration of autoantibodies reactive with the 23 kDa retinal CAR antigen, located within the photoreceptors. Although paraneoplasia antibodies can be reduced by plasmapheresis and immunosuppressants, only questionable results are reported, and controversy persists over consequences of reducing the immune competence of cancer patients. Intravenous immunoglobulin is an alternative treatment option.144a In the setting of cancer, especially with rapid visual loss and normal ERG, the likelihood of carcinomatous meningitis should be considered, as well as the possibility of complications of chemotherapy. Autoimmune retinopathy may occur without evidence of cancer, but with typical photopsias, field depression, ERG abnormality, and sera containing antiretinal antibodies directed against inner plexiform layer, as opposed to CAR.145
CONGENITAL HAMARTOMA SYNDROMES
The “neurophakomatoses” are a diverse group of disorders nosologically related by the presence of hamartomatous lesions, and, indeed, the term “hereditary hamartomatosis” is a more accurate description. However, whereas neurofibromatosis, tuberous sclerosis, and von Hippel-Lindau disease are transmitted with irregular dominance and considerable variation in penetrance, no hereditary basis of Sturge-Weber or angio-osteohypertrophy (Klippel-Trenaunay-Weber) syndrome has been established.
A hamartoma is a tumor of anomalous origin composed of elements normally present in the tissue in which it originates and with a limited capacity for proliferation. The following tumors may be classified as hamartomas: (1) in neurofibromatosis: optic gliomas (see Chapter 6), neurofibromas, and ganglioneuromas; (2) in tuberous sclerosis: retinal and cerebral astrocytomas, cutaneous angiofibromas (“adenoma sebaceum”), rhabdomyomas, and leiomyomas; (3) in von Hippel-Lindau disease: hemangioblastomas of the cerebellum and retina (including optic nerve head) and renal hypernephromas or cysts; (4) in Sturge-Weber disease: facial and choroidal cavernous hemangiomas and meningeal angiomatous malformations; and (5) in Klippel-Trenaunay-Weber syndrome: cutaneous nevi, visceral and limb hemangiomas, and orbitofacial venous varices.
If all disorders with neurocutaneous manifestations are considered, the term phakomatoses (Greek, phakos, “spot,” “birthmark”) is appropriate, and the catalog of “related” disorders becomes cumbersome. “The Phakomatoses,” Volume 14 of Vinken and Bruyn’s Handbook of Clinical Neurology, is extraordinarily complete and serves as a source of detailed clinical descriptions of these diseases.146 Syndromes characterized by vascular hamartomas, that is, retinal-cerebellar angiomatosis (von Hippel-Lindau), and other angiomatous malformations, are discussed in Volume 2, Chapter 17.
TUBEROUS SCLEROSIS
Tuberous sclerosis, so-called because of cerebral tubers (potatoes), is a multiorgan complex that often shows the stigmata of retinal astrocytic hamartomas in epipapillary and parapapillary locations, as well as in the retinal periphery (Fig. 10; see Color Plate 5-1B). These characteristic lesions appear as elevated semitransparent domes in the nerve fiber layer of the retina and may undergo calcification as the patient ages. The calcified hamartomas, when on or near the optic disc, have been termed “giant drusen.” These tumors should not be confused with the much more common drusen (hyaline bodies; see Part II of this chapter, Optic Nerve) within the substance of the nerve head, which are nonhamartomatous lesions and are not characterized by astrocytic hyperplasia. There is no evidence to support the suggestion that hyaline bodies of the optic disc are minor manifestations of tuberous sclerosis.
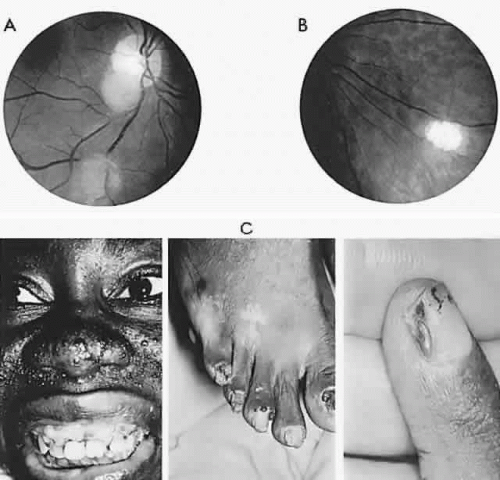 Fig. 10. Tuberous sclerosis. Retinal astrocytic hamartomas in epipapillary, parapapillary, and peripheral sites. A. Superficial translucent lesions through which retinal vessels may be seen. B. Peripheral calcified “mulberry” lesion. C. Cutaneous stigmata include facial fibroma (“adenoma sebaceum”) and periungual fibroma of the toes and fingers. |
 Color Plate 5-1. A. Myelinated nerve fibers. Retina is white, opaque, with feathered edges. B. Calcified astrocytic hamartoma of retinal nerve fiber layer in tuberous sclerosis. C. Hypoplasia of the optic nerve. Disk is small and with pigment rim and surrounding paler ring. Disk vessels appear disproportionately large. D. Inferior crescent. Disk is small and horizontally oval with scleral crescent at lower border. Contiguous inferior fundus sector is hypopigmented and appears albinotic; foveal reflext is indistinct. E. Pseudopapilledema; congenital elevated disk (compare with true papilledema, F). Note absence of central cup, vessels arise at disk apex. Vascular anomalies include excessive number of major disk vessels and multiple bifurcations. Nerve fiber layer does not obscure vessels at disk margins. F. Chronic moderate papilledema (compare with pseudopapilledema in E.) Note retention of central cup, flame hemorrhage at superior border, absence of anomalous vessel pattern, small arterioles are obscured in nerve fiber layer. |
The retinal hamartomas are said to occur in approximately half of patients with tuberous sclerosis. Though rarely symptomatic themselves, they are of great help in establishing diagnosis in the setting of seizures, facial angiofibromas and variable mental retardation. Seizures and EEG abnormalities are present in 80% to 90%, adenoma sebaceum in 80%, mental retardation in 60%, intracranial calcifications in 50%, as well as cardiac rhabdomyomas or hamartomas.147 Because of ventricular obstruction by giant cell astrocytomas, papilledema may develop and consequently visual loss.148 On rare occasions abnormal capillaries on optic disc astrocytomas may abruptly alter vision because of intravitreal hemorrhage.149 Familial occurrence in first-degree relatives should be sought, but some 60% of cases arise as new mutations.
Neuroimaging in tuberous sclerosis demonstrates subependymal (periventricular) nodules, calcifications, and parenchymal hamartomas (cortical tubers); although calcifications are difficult to discern, MRI is considered a better screening procedure.150
NEUROFIBROMATOSIS
Neurofibromatosis embraces at least two disease entities. General neurofibromatosis (NF-1; von Recklinghausen’s disease) is characterized by café-au-lait skin lesions, neurofibromas, schwannomas, optic-chiasmatic gliomas, and Lisch’s nodules on the iris. NF-1 is the most commonly inherited CNS disorder, with an estimated prevalence of 1 in 3000 in Western countries, with gene locus at chromosome region 17q11.2. Lesions of the fundus are rare in neurofibromatosis, although some authors believe that the astrocytic hamartomas typical of tuberous sclerosis also occur in patients with neurofibromatosis. Although there is said to be an increased incidence of medullated retinal nerve fibers, no convincing evidence exists. Multiple gray-brown to yellow, placoid choroidal nevi may be seen at the posterior pole,151 representing a proliferation of Schwann’s cells and occurring in 50% of patients with neurofibromatosis.152 Other stigmata include neurofibroma and neurilemmoma of the orbit and lid, neuronal hamartomas of the iris (Lisch’s nodules) and trabecular meshwork, congenital glaucoma, and sphenoid wing dysplasia with pulsating enophthalmos or exophthalmos.
Neurofibromatosis-2 (NF-2) is an autosomal dominant trait characterized by bilateral acoustic schwannomas associated with other neurofibromas, gliomas, and schwannomas. NF-2 is caused by a mutation in the chromosome region 22q12, with a prevalence rate of only 0.1 in 100,000.147 Cutaneous lesions such as café-au-lait spots and neurofibromas are also found in NF-2. Various cataracts, posterior subcapsular or cortical, are described, as well as small tuberous sclerosis-type retinal nerve fiber and pigment epithelial hamartomas and macular epiretinal membranes, but not Lisch’s iris nodules, as found in NF-1.153 A limited study154 of the behavior of optic nerve gliomas in NF-1 versus those in NF-2 suggests that there is better survival rate with less likelihood of tumor progression in NF-1. In addition, in NF-1 tumors are located more anteriorly, that is, in the orbit and chiasm, than in the optic tract or hypothalamus. Optic gliomas are discussed in Part II of this chapter.
Part II. The Optic Nerve
My lad asked me to look up a word in the French Dictionary and I could not make out the letters because of the bouncing grey dots. The next day I could see peripheral colours but the central zone of vision was covered with what seemed like a grey asbestos mat that occluded all light. The next day saw a further deterioration, loss of colour and general darkening…. In three days all visual response had been lost.
High on the right of my circle of vision, through the murk, I could make out my fingers fluttering after two weeks of blackness. “Hello, Cobalt Blue!” Response to the ophthalmoscope. Strong white light was blue….
It was a thrill to see the cloud and identify its shape at such distance even though that mid-grey was as near as I could get to white…. A friend brought me a plant with red flowers…. “You may have difficulty with reds.” … Another pot of flowers but what fascinated me was the pointillist-like disintegration of each detail … At home. The grey flicks are still there but … I add this ironic homage … “You won’t notice any difference but a doctor could tell.”
Artist Peter MacKarell went blind in one eye and then recovered, no doubt due to optic neuritis.
—New Scientist, February, 1982
The optic nerve, roughly the diameter of thick spaghetti, is subject to a staggering variety of congenital anomalies and acquired disorders. Or perhaps it is even more remarkable that most persons are born without and remain free of optic nerve disease over a long lifetime. From simple myopia, optic neuritis, glaucoma, infarcts, and even its unfavorable location in the crowded skull base, the second numbered cranial nerve is victim to the widest spectrum of developmental malformations and disease processes. Consider the gross anatomic relationships (Fig. 5-11): At the nerve head, the peculiarities of vascular supply and ocular tissue tension (intraocular pressure) are unique; in the orbit, the nerve may be compromised by muscle enlargement or soft tissue tumors; the optic canal is flimsy protection from fractures or sinus inflammations; and the neighborhood relationship with the pituitary gland and arterial circle is clearly an evolutionary disadvantage. Indeed, malfunctions of the optic nerves and chiasm, taken together as the anterior visual pathways, constitute a large portion of all neuro-ophthalmologic diagnostic and management challenges.
 Figure 11. Axial anatomic section of the course of the optic nerves. G, globes; small black arrows just behind globes show the subarachnoid space within the optic nerve sheaths; S, sphenoid, and E, ethmoid air cells; AC, anterior clinoids with dark bone marrow; open black arrows on the canalicular segments of the optic nerves indicate the thin bony medial optic canal wall shared with the sphenoid sinus; white arrow indicates the tuberculum of the planum sphenoidale; asterisk indicates chiasm bordered by carotid arteries, C; P, pituitary stalk; oculomotor nerves, three with open arrows. (Courtesy of Dr. Renate Unsold.) |
CONGENITAL OPTIC DISC DYSPLASIAS AND ANOMALIES
Considerable variation in optic disc size exists, influenced at least in part by refractive error. Megalopapilla, other than those disc variants occurring in myopia or with frank colobomatous malformations, must be exceedingly rare, but micropapilla (disc hypoplasia) is not and warrants inclusion in the diagnostic dilemma of the child with poor vision in one or both eyes. Other congenital disc malformations are accompanied by field defects that may be mistaken for acquired disease, or they are regularly misinterpreted as nerve head swelling.
Among those benign conditions mistaken for new disease is aberrant myelination of the retinal nerve fiber layer on and around the disc, which presents a funduscopic picture of intensely white patches with feathered edges (Fig. 5-12). Said to occur in <1% of ophthalmologic patients, slightly more common in men, and bilateral in 20%, such myelination is anecdotally linked to numerous conditions, including the triad of amblyopia, strabismus, and myopia.1 Although considered a congenital anomaly, retinal nerve fiber layer myelination has been seen to progress in childhood2 or in adult life3 and to disappear following optic nerve damage owing to chronic papilledema,4 Leber hereditary optic neuropathy,5 and other causes.
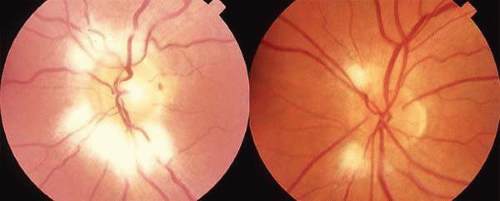 Figure 12. Myelinated nerve fiber layer in two patients. Left. Two feathery white patches adjacent to the optic disc. Right. More extensive and confluent myelination adjacent to the disc. |
Optic Nerve Hypoplasia
Hypoplasia of the optic disc may be unilateral or bilateral, marked or minimal, and associated with good or poor visual function. The condition may occur in isolation or may accompany other ocular or forebrain malformations, with or without developmental delay or endocrine deficiency. An apparent increased prevalence of optic nerve hypoplasia (ONH) may be caused by increased abuse of alcohol and drugs, as suggested by some authors,6,7 but may simply be owing to a heightened awareness of this funduscopic anomaly and its systemic ramifications.
At least two distinct mechanisms have been proposed as the basis for ONH. A deficiency of axon guidance molecules may result in failure of retinal ganglion cells to properly exit into the optic nerve.8 Alternatively, there may be an augmentation of the normal apoptosis (dying back) of retinal ganglion cells owing to toxins or other intrauterine events. The occasional association of ONH with a suprasellar tumor suggests that mechanical disruption may interfere with normal migration of retinal ganglion cells also causing an excessive dying back of axons.9 Prenatal damage to retrogeniculate pathways may also cause segmental hypoplasia, because of retrograde transsynaptic degeneration.10,11 Various potentially teratogenic agents have been associated with ONH in some cases, including phenytoin, quinine, alcohol, lysergic acid, and cocaine. Maternal diabetes has been implicated in some cases, particularly in segmental hypoplasia of the superior portion of the nerve head12,13 (Fig. 5-13). A variant of ONH has been described in patients with periventricular leukomalacia, indicating intrauterine or perinatal hemispheric injury, in which the total size of the optic disc is normal but the cup is unusually large.14
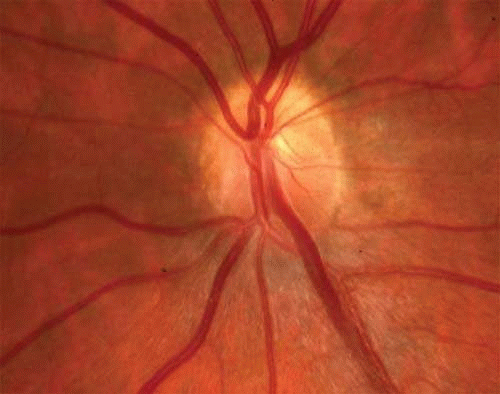 Figure 13. Segmental optic nerve hypoplasia in the offspring of a diabetic mother. Bilateral inferior altitudinal field defects were found on routine screening. The top half of the optic disc is maldeveloped, creating a crescent composed of sclera and retinal elements with absence of nerve fiber layer. The fellow eye had a similar appearance. |
The classic funduscopic appearance in cases of severe ONH may include the double ring sign, the center ring formed by the edge of the true (hypoplastic) disc and the outer ring representing the normal size of the scleral canal (Fig. 5-14). The mismatch between the rings has a variably pigmented appearance and may be difficult to appreciate, especially when examining an uncooperative child. In less severe cases, the nerve is simply smaller than normal, a determination that can be difficult, especially since such observations are influenced by the refractive state of the eye in question. In such cases, fundus diagnosis may be supported by careful side-by-side comparison of disc photographs, or calculation from photographs of the ratio of the disc center-to-fovea distance (DM), to disc diameter (DD); this ratio (DM/DD) is significantly higher in hypoplasia than in normal fundi, a ratio >3.0 being considered diagnostic.15 Otherwise, optic disc dimensions may be assessed using the Goldmann three-mirror contact lens and adjustable slitlamp beam to measure vertical and horizontal disc diameters and applying the formula for area of an ellipse, according to the method of Jonas and Papastathopoulos.16 In eyes with minimal spherical refractive error, Zeki et al17 found that a ratio of disc-to-macula to DD of 2.94 provides a 95% population upper limit, whereas disc hypoplasia has a mean ratio of 3.5.
 Figure 14. A. Severe optic disc hypoplasia, showing a characteristic double ring sign. B. Septo-optic dysplasia: magnetic resonance coronal images. Left. Absent septum pellucidum with a single midline ventricle; arrows indicate prechiasmal optic nerves: L, left nerve is hypoplastic (compare with right, R). Right. At chiasm (open arrow), note the hypoplastic left nerve contribution (arrow). |
Visual acuity in ONH ranges from 20/20 to no light perception,18 and, as a rule, the smaller the disc, the worse is the vision. Visual field defects are variable, including central depression, nasal and temporal wedges and hemianopias, altitudinal loss, and generalized constrictions.
When hypoplasia is bilateral and is accompanied by poor vision and nystagmus, most patients harbor other developmental abnormalities, but such defects occur in only about 20% of unilateral or segmental cases of hypoplasia.19 The congenital syndrome of septo-optic dysplasia (de Morsier syndrome) may be recognized by the clinical triad of short stature, nystagmus, and optic disc hypoplasia. Neuroimaging in such cases demonstrates absence of the septum pellucidum often associated with other hemispheric migration anomalies such as schizencephaly and cortical heterotopia.20 Posterior pituitary ectopia (characterized by absence of the normal posterior pituitary bright spot, an ectopic hyperintense focus in the tuber cinereum and absence of the infundibulum) is often seen as well and is an excellent marker for associated endocrine dysfunction21 (Fig. 5-14B). Developmental and endocrine abnormalities may include neonatal hypotonia, seizures, and prolonged bilirubinemia; growth hormone, corticotropin, and antidiuretic hormone deficiencies; and mental retardation.19 Recognition of these associated endocrine abnormalities is important because deficits may be corrected, resulting in resumption of normal skeletal growth patterns. In addition, Brodsky et al22 reported sudden death in patients with corticotropin deficiency, diabetes insipidus, and thermoregulatory disturbances, aggravated by fever and dehydration.
Neuroimaging, preferably with magnetic resonance imaging (MRI), and endocrinologic assessment are usually recommended for patients with bilateral ONH whereas patients with unilateral or segmental disc hypoplasia, in whom growth and development are normal, can usually be followed expectantly with regular ophthalmologic and pediatric examinations. A trial of occlusion therapy is reasonable for children with unilateral ONH and reduced vision to explore the possible contribution of amblyopia.
True optic nerve aplasia is an exceedingly rare condition, usually found in a unilaterally malformed eye in an otherwise normal individual.23 In rare bilateral cases, there are usually other severe congenital central nervous system (CNS) malformations although one case of an otherwise healthy infant with bilateral aplasia of optic nerves, chiasm, and tracts has been reported.24
Colobomas and Pits
Optic nerve colobomas (Greek, mutilation or curtailment) or pits are congenital malformations that enlarge or distort the nerve head circumference and assume several forms (Fig. 5-15; also Volume 2, Chapter 13, Fig. 4): enlarged discs with deep excavation; enlarged, relatively round discs filled with retained embryonic glial and vascular remnants, at times projecting forward as a funnel (morning glory syndrome of Kindler); discs posteriorly displaced within excavated peripapillary staphylomas; dysplastic, excavated, vertically oblong discs contiguous with retinochoroidal colobomas, located especially inferiorly; and slightly enlarged, irregular discs containing pits within the borders of the nerve head (discussed later). In most instances, the expanded peripapillary area is irregularly pigmented and is crossed by numerous anomalous vessels (Fig. 5-15). These conditions probably represent different degrees of dysplasia in the spectrum of optic nerve malformations possibly related to faulty closure of the embryonic ventral (fetal) fissure of the optic stalk and cup25 and are sometimes referred to as cavitary disc anomalies. Gopal et al26 categorized several variants of disc position and morphology in the spectrum of coloboma, most discs being included in the retinochoroidal defect itself. Rare instances of coloboma show momentary dynamic changes in the surrounding peripapillary staphyloma, described as contractile or pulsatile, and possibly related to mesodermal fat or smooth muscle replacing the optic meninges.27
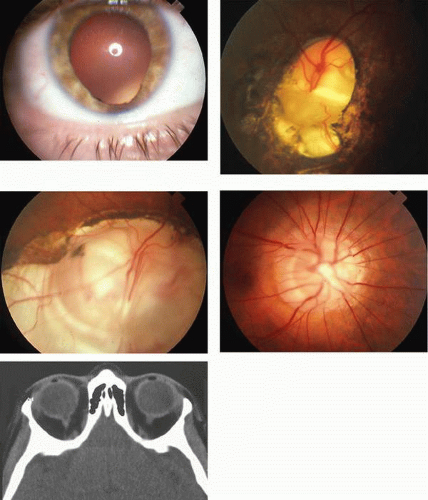 Figure 15. Cavitary disc anomalies. A. Iris coloboma with a typical keyhole appearance. B. In the same individual, an optic disc coloboma showing marked excavation of the inferior portion of the disc with adjacent staphyloma. C. More extensive optic disc coloboma in a different patient. Disc excavation is partially filled in by glial tissue. D. A dysplastic disc with anomalous vasculature and peripapillary pigmentary disturbance, which on CT (E) can be seen to be markedly excavated. |
Colobomas and related cavitary disc anomalies may occur as a dominantly inherited trait,28 but most are unilateral and sporadic, especially the morning glory type.27 Simple disc colobomas may be accompanied by various systemic disorders,6 including Aicardi syndrome, CHARGE association (congenital heart disease, choanal atresia), oculoauricular dysplasia (Goldenhar), linear sebaceous nevus, Goltz focal dermal hypoplasia, and Walker-Warburg syndrome. Colobomas have also been associated with orbital cysts and arachnoid cysts29 and with Dandy-Walker malformation.30 Unilateral coloboma has been reported with vascular lesions including carotid occlusion, moyamoya collateralization with dolichoectasia, and absent ophthalmic artery.31
Of great clinical importance is the association of disc malformations, especially morning glory,32 with congenital forebrain anomalies, including basal encephaloceles. Herniated brain tissue may present as pulsating exophthalmos (sphenoorbital encephalocele, most commonly in neurofibromatosis), hypertelorism with a pulsatile nasopharyngeal mass (transsphenoidal encephalocele), or a frontonasal mass, with or without hypertelorism (frontoethmoidal encephalocele) or other midfacial malformation. The physical findings of transsphenoidal or transethmoidal basal encephalocele are as follows:
| Midline facial anomalies |
| Broad nasal root |
| Hypertelorism |
| Midline lip defect |
| Wide bitemporal skull diameter |
| Cleft palate |
| Nasopharyngeal mass |
| Midline pharyngeal space |
| Pulsatile |
| Symptoms of nasal obstruction |
| “Nasal polyp” (true polyp rare in infancy) |
| Hypopituitarism/dwarfism |
| Ocular |
| Congenital disc anomalies (colobomatous dysplasias) |
| Chiasmal field defects, poor vision |
| Exotropia |
Neuroimaging of the skull base, the chiasm, and the inferoanterior brain structures affirms the diagnosis. Biopsy or attempted resection of such posterior nasopharyngeal masses should not be attempted because they may contain vital structures including visual pathways and the hypothalamic-pituitary system. Patients with congenital disc malformations may complain of transient obscurations of vision lasting seconds to minutes, but the mechanism of visual disturbance is unknown.
Pits of the optic disc are usually definable ophthalmoscopically as intrapapillary pearly gray dimples or slits containing filmy pale glial material, located typically just within the scleral rim of the disc margin, extending about 2 clock hours or one third of the disc diameter. The disc border is frequently distorted and may be highlighted by contiguous mild pigment epithelial changes (Fig. 5-16), and cilioretinal vessels may traverse the depression.33 Pits usually occur singly in a temporal location, less frequently centrally, or in an inferior, superior, or nasal quadrant, but they may be multiple and bilateral in perhaps some 15% of patients. They are rarely familial, but an autosomal-dominant inheritance pattern is possible. Indeed, Ragge et al34 suggested that there is evidence that cavitary anomalies of the disc form a spectrum ranging from pits to colobomas, and the existence of persons with pits in one eye and contralateral coloboma implies that these anomalies are variations of the same genetic or environmental insult; moreover, pedigrees are reported that contain the various phenotypic expressions of the cavitary anomalies, some related to mutations of the PAX2 gene.35
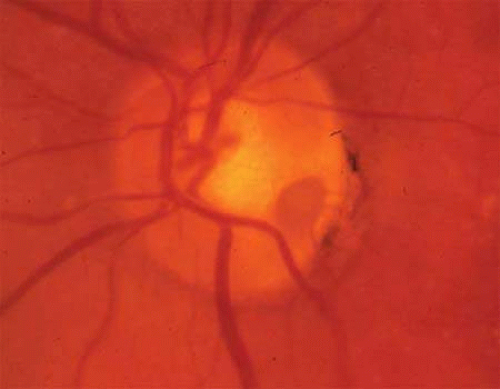 Figure 16. Optic pit. Typical grayish, oval depression in the inferotemporal aspect of an enlarged disc. A small cilioretinal artery is seen emerging from the pit. |
The association of temporal pits with serous detachment of the macula, and consequent diminished acuity, is well known, with an incidence perhaps in 50% of patients,36 usually presenting in young adulthood. Otherwise, stable visual field changes take the form of dense nerve fiber bundle defects that extend from the blind spot, especially toward fixation in the papillomacular zone. In contrast to congenital disc pits, acquired pits, especially at the inferior disc margin, are part of the spectrum of glaucomatous excavation, perhaps more frequent with normal ocular tension, typically with dense field depressions in the central portion of the visual field.37 Pulsatile communication of fluid between the vitreous cavity and a retrobulbar cyst via an optic pit has been demonstrated.38
Dysversions and Crescents
Field defects associated with congenital dysversions of the optic disc (tilted discs, situs inversus) and accompanying depigmented peripapillary crescents may be confused with the bitemporal hemianopia of acquired chiasmal lesions. The most common variety of crescent is that located inferiorly (inferior conus, Fuchs coloboma), first described by Fuchs in 1882 (Fig. 5-17A). Not only is the disc hypoplastic, ovoid, and vertically truncated, but also the fundus in the sector contiguous to the crescent takes on a semialbinotic or tigroid appearance because of hypopigmentation of pigment epithelium and choroid. As a rule, inferior crescents are associated with moderate myopia with astigmatism (usually manifest in the same axis as the dysversion of the disc, that is, between 90° and 110° in right eyes and between 90° and 70° in left eyes), slightly reduced corrected visual acuity, and abnormal foveal reflex. Because the inferonasal retinal quadrant is involved most frequently, relative superior temporal field defects are found (Fig. 5-17B), which may simulate bitemporal hemianopia owing to chiasmal disease.39 Some or all of this visual field defect is a consequence of regional myopia localized to the inferonasal retina, as evidenced by its correction using additional minus lenses for this portion of the test. The superior temporal defect of tilted disc syndrome can usually be distinguished from a true bitemporal defect by its failure to align with the vertical meridian. Riise40 provided an excellent monograph on this entity. Disc abnormalities in craniofacial disorders, including hypertelorism and Crouzon and Apert syndromes, show a spectrum of forms: simple pallor, tilt and inferior conus, and coloboma, some with widespread fundus hypopigmentation.
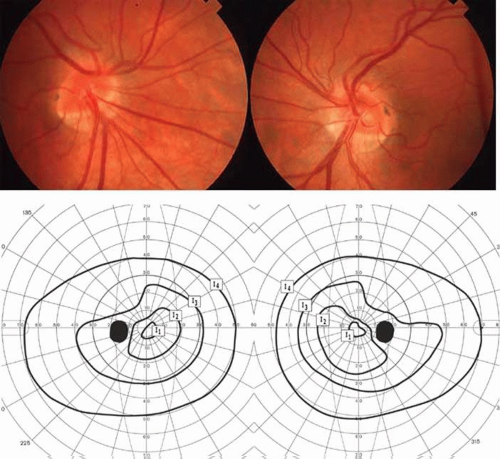 Figure 17. Tilted discs (Fuchs coloboma). A. Both discs are anomalous, showing hypoplasia of the inferonasal aspect with a large inferonasal scleral crescent. B. Goldmann perimetry shows a characteristic pseudobitemporal defect, which fails to align along the vertical meridian. This feature is helpful for distinguishing this from the true bitemporal defect associated with chiasmal compression. |
Anomalous Disc Elevations: Pseudopapilledema and Hyaline Bodies
Anomalous elevation of the optic nerve head, with or without ophthalmoscopically detectable hyaline bodies, is a major cause of unnecessary alarm and misdirected diagnostic procedures. Because this funduscopic appearance resembles acquired disc swelling, including papilledema of raised intracranial pressure, patients are often subjected to extensive and invasive neurodiagnostic tests including scans and lumbar puncture for innocent headaches, vertigo, or more trivial symptoms. Nowhere in neuro-ophthalmology is funduscopic differentiation more critical; an accurate diagnosis of pseudopapilledema can prevent a multitude of unnecessary tests.
Congenitally elevated discs have also been termed pseudopapilledema, but, when possible, more specific funduscopic characteristics should be described (e.g., intrapapillary hyaline bodies, simple hyperopia, persistent hyaloid tissue). Most cases of anomalous elevation are associated with hyaline bodies of the nerve head, also termed optic disc drusen.
Disc drusen are a common congenital anomaly, occurring in up to 2% of the population41 and inherited in an autosomal-dominant fashion with irregular penetrance.42 Drusen are composed of acellular laminated concretions, often partially calcified, which are related to accumulation of axoplasmic derivatives of degenerating retinal nerve fibers. The basis for this neuronal degeneration is unclear. It has been suggested that an abnormally small scleral canal produces mechanical compression of optic nerve axons.43,44 Measurements of the scleral canal in a group of 25 patients with disc drusen, however, failed to support this theory.45 Alternatively, a primary defect in retrograde axonal transport may cause cell death followed by extrusion of intra-axonal contents.43,46 Finally, a congenital dysplasia of disc vasculature may be responsible.47 In support of the latter theory, Antcliff and Spalton47 found anomalous disc vessels in 57% of eyes of family members of individuals with disc drusen.
Hyaline bodies slowly become more evident as they enlarge toward the disc surface and margins. Thus, anomalously elevated discs in children usually do not show ophthalmoscopically detectable hyaline bodies (said to be buried), which insidiously emerge by the early teens.48 Hoover et al48 reported first evidence in one or both eyes at a mean age of 12.1 years in 40 children. Visible drusen have been reported in children as young as 3 years.49 The occurrence of overt hyaline bodies in parents of children with anomalously elevated discs, but without apparent hyaline bodies, attests to both the progressive and the heredofamilial nature of this disorder. Indeed, some family members have visible hyaline bodies, whereas others have only elevated discs (Fig. 5-18). Examination of family members may be helpful when the distinction between true papilledema and pseudopapilledema is in doubt. In addition to this slow growth of drusen over time, the overlying retinal nerve fiber layer also becomes progressively thinner. This decrease in nerve fiber layer is demonstrable both funduscopically and with optical coherence tomography (OCT).50
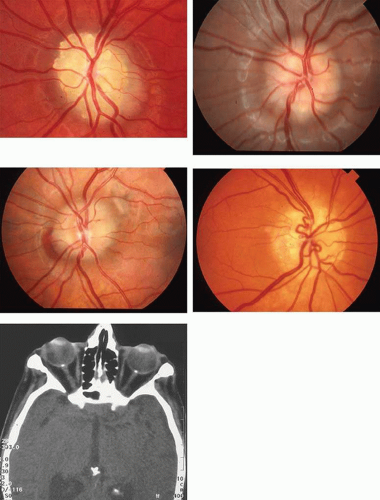 Figure 18. Optic disc drusen. A. Multiple surface drusen. B. Drusen barely visible just under the surface with scalloped disc margins. C. Buried drusen with a characteristic deep, crescentic, peripapillary hemorrhage. D. Drusen with anomalous vasculature and peripapillary pigmentary changes. E. CT in a patient with bilateral optic disc drusen showing bright calcification within the substance of each disc. |
Although one disc may be more elevated than the other, both with or without apparent hyaline bodies, drusen are commonly bilateral. Lorentzen42 found ophthalmoscopically visible hyaline bodies in 73% of cases and Rosenberg et al51 in 69%. Mustonen52 studied 184 patients and found hyaline bodies were bilateral in 66.9%, strictly unilateral in 25.5%, and evidenced by disc elevation without hyaline bodies in the contralateral eye in 7.6%. There is no significant relationship between hyaline bodies and refractive error.51 With the infrequent exception of retinitis pigmentosa53 angioid streaks (with and without pseudoxanthoma elasticum),54 and Alagille syndrome,55 there appears to be no statistically significant association of hyaline bodies with the numerous and diverse ocular and neurologic disorders with which they have been described.51
Although most patients with disc drusen are asymptomatic, 71% to 75% have peripheral visual field defects.56 Field defects usually take the form of blind spot enlargement, arcuate or other nerve fiber bundle patterns, or irregular peripheral contraction56 (Fig. 5-19). These deficits typically progress exceedingly slowly, with a predilection to involve the inferior nasal quadrant. Because enlarged blind spots occur in both pseudopapilledema and true papilledema, this finding is of no differential diagnostic significance. The central field is generally spared.
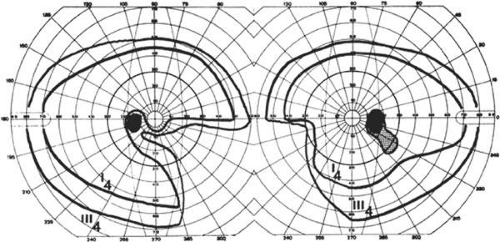 Figure 19. Visual field defects with optic disc drusen. There is an inferior-nasal defect in each eye consistent with nerve fiber bundle defects. In the right eye, the peripheral field is preferentially affected. |
In occasional cases, patients with optic disc drusen experience an abrupt decline in vision owing to hemorrhage or ischemia.57 Hemorrhage may be related to choroidal neovascular membrane formation in some cases, which may occur in children.58 Sudden loss owing to ischemia usually takes the form of anterior ischemic optic neuropathy (AION).59 In cases of AION with drusen, the clinical features are similar to those of nonarteritic AION except for a younger age at onset and a more favorable visual outcome.60 Loss of central field, that is, diminished acuity, should not be attributed to disc drusen unless it is caused by hemorrhagic complications or ischemic disc infarction or is associated with profound loss of peripheral field.61
It is not uncommon to elicit a history of transient obscuration of vision in patients with hyaline bodies,42 and this symptom may further serve to confuse the clinical differentiation from true papilledema. These episodes may last seconds to hours, and vision may be profoundly affected during the episode.62 Sarkies and Sanders63 have documented the extraordinary history of 26 years of recurrent episodic visual loss associated with disc drusen, perhaps related to anomalous vasculature.
Ophthalmoscopic criteria that distinguish between true and pseudopapilledema are as follows:
The central cup is absent; the disc diameter tends toward small.
Vessels arise from the central apex of the disc.
Anomalous branching of vessels occurs; an increased number of major disc vessels is noted; venous pulsation is present.
The disc may be transilluminated with a focal light source of the ophthalmoscope or slit beam, with a glow of hyaline bodies when present.
The disc margins are irregular with derangement of peripapillary retinal pigment epithelium.
Superficial capillary telangiectasia is absent.
Hemorrhages are usually absent; when present they are usually deep and crescentic (Fig. 5-18C).
No exudates or cotton-wool spots are present.
If difficulty in fundus diagnosis persists, the following rules may prove valuable: A spontaneous venous pulsation militates strongly against papilledema of increased cerebrospinal fluid (CSF) pressure; if the patient is otherwise thriving, it is probably not papilledema; computed tomography (CT) scan and ultrasonography can reveal buried hyaline bodies (Fig. 5-18E).
HEREDODEGENERATIVE OPTIC ATROPHIES
Among the causes of insidious, bilateral, and symmetric loss of central vision must be considered the heredodegenerative optic atrophies. Although it is seemingly a simple task to uncover familial incidence, in many cases such patterns cannot easily be established or are confounded by variations in phenotypic expression. Optic abiotrophies may occur as monosymptomatic isolated bilateral central visual defects, or they may accompany other nervous system degenerations involving motor, sensory, and auditory function. Optic atrophy also evolves secondarily in heritable storage disorders, in which accumulation of abnormal material in retinal ganglion cells results in consecutive disc pallor (e.g., Tay-Sachs disease; Volume 2, Chapter 5, Part I). Retinitis pigmentosa and other retinal dystrophies, including Leber congenital amaurosis, show variable degrees of optic atrophy, but the primary disorder is the retinal degeneration. Electroretinography (ERG) is a useful tool for making this distinction.
Simple or complicated optic atrophies occur in various patterns of transmission and with graded symptomatology, such that a vast and heterogenous literature has accrued. Table 5-6 is an attempt at pragmatic clinical classification. Historically, cases were grouped based on phenotypic expression. Recent advances in genetics have allowed a more accurate basis for classification.64
TABLE 6. Heredofamilial Optic Atrophies | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
Recessive Optic Atrophy
Simple (Isolated)
Isolated optic atrophy of recessive inheritance is rare. Some previously described cases were probably incorrectly classified, and in fact its very existence has been called into question.65 Reported cases have presented in infancy as severe visual loss with nystagmus. A first locus for isolated recessive optic atrophy has recently been mapped to 8q21-q22 in a large consanguineous family of French origin.66
Complicated (Associated with Other Neurologic Abnormalities)
First described in 1909, infantile recessive atrophy, or Behr syndrome, consists of optic atrophy associated with variable mild mental deficiency, spasticity, hypertonia, and ataxia. Although all of Behr’s original patients were male, subsequent reports have indicated no sex predilection. The disorder has its onset in childhood (1–9 years) and stabilizes after a variable period of progression.67
A disorder that is phenotypically similar to Behr syndrome has been identified in several Iraqi-Jewish pedigrees and mapped to chromosome 19q13.2–13.3.68 It is termed Costeff syndrome or OPA3, and affected individuals have been found to have elevated urinary excretion of 3-methylglutaconic acid.69 Some cases of propionic acidemia have been associated with similar clinical findings.70 Behr syndrome is likely heterogeneous, perhaps representing a transitional form between simple hereditary optic atrophy and the hereditary cerebellar ataxias of the Marie type.
Autosomal recessive optic atrophy has also been reported in association with axonal motor and sensory neuropathy,71 with familial dysautonomia,72 and in a rare condition consisting of progressive encephalopathy, subcutaneous limb edema, hypsarrhythmia, and optic atrophy, termed the PEHO syndrome.73,74
Wolfram Syndrome and Juvenile Diabetes
The association of early childhood–onset optic atrophy with diminished vision and juvenile diabetes is known as Wolfram syndrome, and is recalled by the mnemonic DIDMOAD (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness) (Fig. 5-20A).75 The disc pallor is not directly related to degree of diabetic retinopathy and, indeed, is found even without background retinopathy. Lessell and Rosman76 reported an experience of nine cases that included other associated manifestations, including ptosis, ataxia, nystagmus, seizures, mental retardation, abnormal ERG, elevated spinal fluid protein and cells, and small stature. Other systemic anomalies include ureterohydronephrosis and neurogenic bladder. In the United Kingdom, the prevalence rate is 1/770,000, with median age at presentation of diabetes at 6 years and optic atrophy at 11 years; cerebellar ataxia and myoclonus tend to evolve in the fourth decade, and median age at death is 30 years (range, 25 to 49 years).77
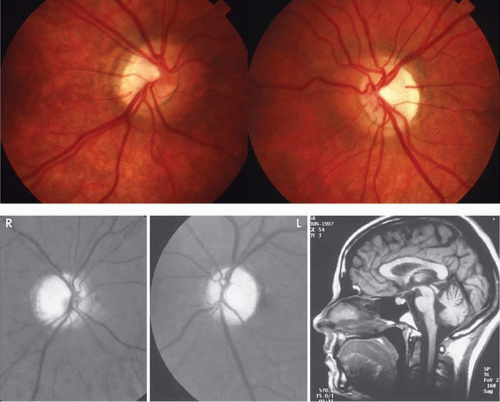 Figure 20. Wolfram syndrome (optic atrophy and diabetes mellitus). A. There is marked bilateral optic disc pallor most pronounced temporally. The discs also have a somewhat tilted configuration, especially the right. B. MRI shows marked atrophy of cerebellum, brainstem, and spinal cord. |
Neuropathologic examination78 has disclosed the following: atrophy of olfactory bulbs and tracts, optic nerves, and chiasm; loss of neurons in small cell layers of the lateral geniculate; atrophy of the superior colliculus; fiber loss in the cochlear nerve and cochlear nuclei; olivopontocerebellar atrophy; and pyramidal tract demyelination. Other neurologic features include central apnea and respiratory failure, startle myoclonus, axial rigidity, and Parinaud syndrome.79 MRI may show striking brain stem atrophy, especially of the pons and midbrain (Fig. 5-20B).
In several families with Wolfram syndrome, the gene was localized to the short arm of chromosome 4 (4p16l6)80; however. multiple point mutations and deletions have subsequently been identified at this locus.81 The fact that many of the abnormalities in Wolfram syndrome are common in patients with mitochondrial disorders has led to speculation that a mitochondrial defect might be responsible for this condition. Indeed, rare patients with Wolfram syndrome have been found to harbor the 11778 DNA mutation associated with Leber hereditary optic neuropathy (LHON) (discussed later),82 and other studies have shown a preponderance of two major mtDNA haplotypes in Wolfram patients that are also overrepresented in patients with LHON.83 It has been proposed that a particular locus on chromosome 4p16 may be a predisposing factor for the formation of multiple mtDNA deletions.84 For these reasons, the Wolfram genotype may not be homogeneous.
To reiterate, the simple autosomal recessive optic atrophies are not common, and patients previously categorized as having recessive congenital or infantile optic atrophy may indeed have suffered from retinal dysplasias or other unrecognized hereditary patterns. Of the more complicated situations in which optic atrophy is associated with clinical manifestations in organs unrelated functionally or embryologically, the possibility of mitochondrial defects is now strongly implicated.
Dominant Optic Atrophy
The monograph on dominant optic atrophy (DOA) by Kjer85 published in 1959 was an important milestone in nosologic analysis of the heritable optic atrophies, defining the clinical parameters and providing further evidence for distinction from Leber hereditary optic atrophy, with which it had previously been confused. Damien Smith86 provided an additional admirable review of dominant optic atrophy and defined diagnostic criteria and clinical variants, emended here with data from a study of 21 families from the Genetic Clinic of Moorfields Hospital87: (i) dominant autosomal inheritance; (ii) insidious onset between the ages of 4 and 8 years; (iii) moderately reduced visual acuity, from 20/30 to 20/70, but rarely so poor as 20/200, with some considerable asymmetry of acuities possible, and modest deterioration with age; (iv) temporal pallor of optic discs, at times with temporal sectoral excavation, and striking thinning in the papillomacular nerve fiber layer (Fig. 5-21); other discs show diffuse atrophy; (v) centrocecal enlargement of the blind spot or midzonal temporal depression with a predilection for the superior-temporal field, at times mimicking temporal central hemianopia (Fig. 21); (vi) full peripheral fields but elevated threshold for motion detection; (vii) acquired blue-yellow dyschromatopsia, which is pathognomonic when present (about 80%), but also with nonspecific or mixed color-confusion axes; and (viii) reduced amplitude and delayed evoked potentials and reduced N95 component of pattern ERG.
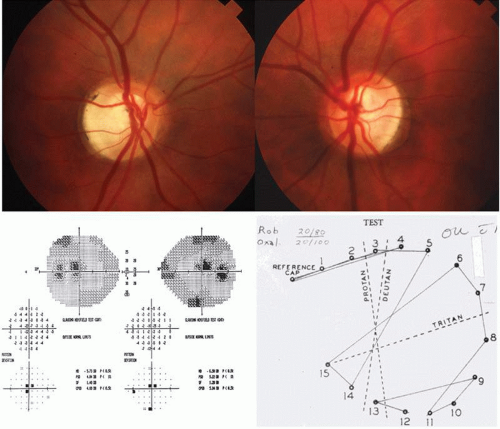 Figure 21. Dominant optic atrophy (OPA1 mutation). A. Both discs show marked temporal pallor and excavation. B. Visual fields show cecocentral depressions mimicking chiasmal disease. C. Plot of D-15 color vision test shows an error pattern aligned with the tritan axis. |
Many patients are ignorant of the familial nature of their disease and, indeed, had themselves not been aware of visual defects,88,89 attesting to the insidious onset and only mildly progressive nature of visual dysfunction in this disorder. Evidence of disease progression is derived from studies of groups of patients segregated by age rather than by the clinical course of individual patients. From Kjer’s data and that of the Moorfields series, patients younger than 15 years of age did not show vision worse than 20/200, whereas 10% of patients 15 to 44 years old, and 25% of patients 45 years and older, had visual function <20/200; none of Kjer’s patients had vision reduced to hand-motion or light-perception levels, although the Moorfields data do include such low levels.
Genomic DNA linkage mapping in patients with DOA has identified various mutations in the OPA1 gene on 3q27–3q28, encoding a mitochondrial dynamin-related GTPase.90,91 Between 30% and 90% of DOA families have been found to harbor over 90 different missense and nonsense mutations, deletions, and insertions in a gene within this region, now designated as the OPA 1 gene.91 Because of this large number of different mutations responsible for the same phenotypic expression, genetic testing is not possible except in those cases in which the specific mutation in a particular family has been identified. The product of the OPA1 gene is targeted to mitochondria and influences function in various ways.92 Loss of the OPA1 protein leads to fragmentation of mitochondrial cristae, release of cytochrome c, and eventual cell apoptosis.91 The OPA1 gene influences mitochondrial function in other ways as well, as evidenced by more widespread neurologic manifestations in some families with OPA1 mutations.93 Amati-Bonneau et al94 have proposed the term “OPA1-plus” syndrome for such families, encompassing progressive external ophthalmoplegia, peripheral neuropathy, ataxia, and deafness in addition to optic atrophy. Other mutations have been identified as causing dominantly inherited optic atrophy without other neurologic deficits, including OPA4, OPA 5, and one mapping to the OPA 3 locus that produces optic atrophy and cataract.95
In addition to the isolated optic atrophy of DOA, there are rare cases of dominantly transmitted optic neuropathy with other deficits. C.S. Hoyt88 reported two families with DOA plus sensorineural hearing loss and nystagmus, and Grehn et al96 documented a similar family with DOA, hearing loss with mutism, no nystagmus, and with normal ERG. Twenty-three members of a large kindred97 demonstrated progressive optic atrophy with hearing loss beginning in the second decade, ataxia, variable a- and b-wave ERG amplitude reductions (without pigmentary retinopathy), ptosis, and moderate gaze palsies. Associated syndromes include a dominant form of relatively asymptomatic hereditary motor and sensory neuropathy (HMSN type IV) with optic atrophy.98
DOA is the most common heredofamilial simple (monosymptomatic) optic atrophy. Visual dysfunction in this disorder is usually considerably milder than in either Leber hereditary optic neuropathy (LHON) or any form of recessive optic atrophy. As a rule, progression is minimal, and prognosis good, but, in some instances, relatively rapid deterioration may occur, even after years of stable visual function. The degree of visual dysfunction is extremely variable, even within families, and it is important to point this out to families when providing genetic counseling for this disorder88,99 (Fig. 5-22).
 Figure 22. Dominant optic atrophy (OPA1 mutation). A. Family H pedigree. Propositi (P): 20/70 (6/21) and 20/100 (6/30) acuity and tritanopia. Father (f): 20/40 (6/12) and 20/50 (6/15) acuity and mild unclassified dyschromatopsia. Grandmother (g): 20/30 (6/9) and 20/40 (6/12) acuity and tritanopia. Cousin (c): 20/70 and 20/100 acuity and undetermined color defect. Arrows indicate persons examined. Other family members (stippling) have reportedly poor vision. B. Visual field in dominant optic atrophy. Note cecocentral scotomas and superotemporal depression of internal isopters I-2,1, which simulates the bitemporal pattern of chiasmal interference. |
Leber Hereditary Optic Neuropathy
In 1871, Leber described a nosologically distinct hereditary form of optic neuropathy that now bears his name. Identification of the mutations responsible for this disorder has allowed for more accurate characterization of the clinical profile.100 Leber hereditary optic neuropathy (LHON) is characterized by sudden and severe loss of visual acuity associated with large, dense central scotomas, sequentially bilateral, occurring mostly in the second and third decades of life. Men account for 80% to 90% of cases in most pedigrees.101 The disorder is inherited strictly in the maternal line (Fig. 5-23) but with incomplete penetrance, and affected males do not transmit the trait. Although most patients with LHON are otherwise healthy, some show cardiac conduction defects and others101,102 have major and minor neurologic abnormalities including dystonia, spasticity, ataxia, encephalopathic episodes and psychiatric disturbances, and a syndrome mimicking multiple sclerosis (MS). The degree of symmetry, high rate of failure to remit, and painless onset serve to distinguish most cases of Leber optic neuropathy from the retrobulbar neuritis of MS. Nonetheless, MRI is appropriate to rule out demyelinative disease In contrast to patients with optic neuritis, T2-weighted fast spin echo and postcontrast T1-weighted MRI sequences do not show signal changes in the optic nerves in the acute stage of LHON.103
 Figure 23. Leber hereditary optic neuropathy. A. Family B pedigree. Three young sons with useful vision in one of six eyes. Mother, age 28 years, suffered acute bilateral loss of vision at age 14 years, with slowly progressive recovery over 6 years. Maternal uncle, age 74, suffered acute bilateral loss of vision at age 18. Note oblique transmission in the female line. Arrows indicate persons examined. B. Typical dense central field defects. |
During the acute phase of visual loss, the nerve head often appears hyperemic and swollen, as do the dense arcuate retinal nerve fiber bundles above and below the disc, accompanied by tortuosity of large and small peripapillary vessels (Fig. 5-24). Variable arteriolovenular shunting is best demonstrated by fundus fluorescein angiography (Fig. 5-25). Nikoskelainen et al104,105 demonstrated that this characteristic peripapillary microangiopathy occurs also in the presymptomatic phase of involved eyes and in many asymptomatic offspring in the female line. Increased peripapillary capillary shunting and disc hyperemia herald the acute phase of visual loss; slowly, marked disc pallor evolves. Peripapillary and subhyaloid hemorrhage is quite rare. Thus, telangiectatic microangiopathy is a hereditary marker and signifies increased risk of acute optic neuropathy, with a phase of visual loss triggered by as yet unclear environmental or metabolic factors. It should be noted that exhibiting this characteristic funduscopic appearance does not necessarily predict future visual loss. The characteristic thickening of the nerve fiber in LHON has been demonstrated with OCT both in affected individuals106 and in unaffected carriers.107
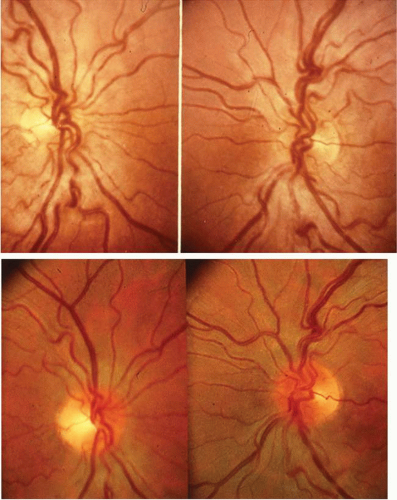 Figure 24. Leber hereditary optic neuropathy. A. Optic discs in a 6-year-old boy with LHON prior to visual loss, showing thickening of the peripapillary nerve fiber (pseudoedema). B. Optic discs of the same individual at age 21, now with decreased vision in the right eye. The right disc now shows temporal pallor; the left disc is still hyperemic but not pale. |
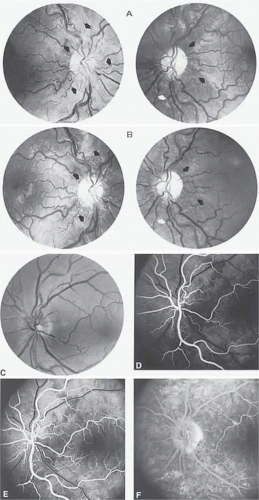 Figure 25. Fluorescein angiography in LHON. A. Left optic disc of a 17-year-old boy with Leber hereditary optic neuropathy. B–D. Fluorescein angiography demonstrates peripapillary microangiopathy, with mildly dilated retinal arterioles (B), capillary shunts in the arteriovenous phase (C). The late phase (D) shows no fluorescein leakage from disc tissue. |
Transmission is strictly maternal because the mother’s egg is the sole provider of mitochondrial (mt) DNA. In LHON, a defect in mtDNA impairs the organelle’s capacity to manufacture adenosine triphosphate, thus causing a failure of cellular energy production. Three specific point mutations account for at least 90% of cases of LHON: at mtDNA nucleotide positions 11778 (accounting for 69% of cases), 3460 (13% of cases) and 14484 (14% of cases).101 Several other mtDNA mutations have been described, but each accounts for only a few pedigrees worldwide. The use of both restriction endonuclease SfNI and Mae III marker sites effectively eliminates the possibility of false-positive test results.108 Instances of apparently nonfamilial (singleton) cases are not rare. Thus testing for LHON is appropriate in patients with undiagnosed chronic or acute but bilateral optic neuropathies even in the absence of a family history of the disease.
Age at onset is usually in the second or third decade, but cases ranging from 2 to 80 years have been reported. Asymmetry of onset is difficult to assess because of patient subjectivity, but some interval of weeks to a few months would appear to be the rule. According to Nikoskelainen et al,104 40% of patients are unsure of the interval between eyes, but second eye onset was <2 months in 23%, from 2 to 6 months in 32%, and >6 months in only 6%. Newman et al109 reported simultaneous onset in 55% of patients with 11778 mutation and an intereye interval of 1.8 months, whereas in the British study,108 visual loss was simultaneous in 22% and sequential in 78%, with a median intereye delay of 8 weeks. Individual variations in severity and tempo may be confusing, but entirely unilateral cases are distinctly rare104,109,110; One case with the 11778 mutation was considered unilateral for 16 years.104 Of interest, monozygotic (identical) twins homoplastic for mitochondrial mutations 4216, 13708, and 11778 are reported, one with typical visual loss at age 34 years, the second with normal visual function and unremarkable optic discs after more than 6 years of observation.111
In most patients with LHON, visual loss is severe and permanent. Occasional recovery, however, has been well documented, in many cases taking the form of a small area of clearing within a large central scotoma, sometimes termed a fenestrated scotoma. The likelihood of such recovery is related to the specific mutation. Among patients with the 11778 mutation only 4%112 to 22%113 were found to have spontaneous recovery compared to 37% to 65%110,113,114 of those with the 14484 mutation. When recovery does occur, it is usually a year or more after onset. The presence of disc pallor does not rule out the possibility of some future improvement of vision. A younger age at onset may convey a better prognosis for recovery110; not all studies confirm this.113
There are no known effective therapeutic measures, medical or surgical, although on theoretical grounds, succinate and coenzyme Q, cofactors for normal mitochondrial function, may be beneficial. A trial of brimonidine in a small group of patients with unilateral visual loss caused by LHON failed to prevent visual loss in the fellow eye.115 The possible role of exogenous trigger factors has been investigated with variable findings. Cases of acute visual loss in individuals harboring the LHON mutation shortly after starting highly active antiretroviral therapy (HAART) for HIV/AIDS suggest a similar mechanism, in which an exogenous factor (in this case medication) produces oxidative stress in a system with poor reserves.116,117 It is reasonable to advise affected individuals and carriers to avoid potential toxins such as tobacco, alcohol, and chronic solvent exposure. Genetic counseling for individuals with this disorder should include the fact that all offspring of women harboring this mutation will inherit the condition, but only 25% of at-risk males and 5% of at-risk females actually experience visual loss.115
Neurodegenerative Diseases
There is considerable overlapping of syndromes variously combining progressive degeneration of cerebellar and pyramidal systems, deafness, and optic atrophy. To add to the nosologic confusion, familial progressive polyneuropathies of the Charcot-Marie-Tooth type have occasionally been associated with optic atrophy.118 The association of optic atrophy with spinocerebellar degenerations, familial bulbospinal neuronopathy,119 cerebellar ataxia with sensorineural deafness (CAPOS syndrome),120 and familial dysautonomia72,121 is also well established. Optic nerve dysfunction, although not necessarily symptomatic, is documented in Friedreich-type and hereditary spastic ataxia.122 As previously noted, optic atrophy associated with neurologic signs may take the form of an acute optic neuritis, at least superficially resembling Leber disease, or may occur as a recessive optic atrophy in childhood, associated with ataxia (Behr syndrome). Other ocular findings associated with cerebellar ataxia include ophthalmoplegia with slow saccades, retinal pigmentary degenerations with primary or consecutive optic atrophy.
This frequent intermingling of spinocerebellar degenerations, heredoataxias, motor and sensory neuropathies, deafness, and optic atrophy strongly suggests a complex genetic continuum in which finite distinctions are not yet possible. Transmission may be dominant, as in Charcot-Marie-Tooth polyneuropathy, some cases of Friedreich ataxia, and in CAPOS syndrome, or recessive, as in most cases of Marie disease, or may not conform to strict mendelian rules, or as yet disclosed mitochondrial mutations.
ACQUIRED OPTIC NERVE DISEASE: AN OVERVIEW
In neuro-ophthalmologic practice, many patients are victims of lesions of the prechiasmal visual pathways. These disorders are both varied and numerous and therefore constitute a common diagnostic challenge. The clinical distinction of optic neuropathies from maculopathies and other retinal disorders is elaborated in Volume 2, Chapter 2, and in Part I of this chapter.
Clinical Characteristics
The typical and characteristic symptomatic deficits of visual function with optic nerve disease may be summarized as follows:
Monocular deficits are the rule unless, of course, both optic nerves are involved. Hereditary atrophies and toxic-nutritional neuropathies are bilateral, but they may be asymmetric. Optic neuritis is occasionally bilateral and simultaneous, most frequently in childhood.
Defects in central field function include diminished acuity, desaturation of color perception, a sense of reduced brightness, and sluggish direct pupillary light reaction.
Field defects include central depression and nerve fiber bundle defects. Altitudinal defects are more usually vascular in origin. Optic disc disease (e.g., glaucoma, ischemic neuropathy, hyaline bodies) typically shows inferior nasal predilection.
The appearance of disc pallor depends on the nature of the offending lesion, the time interval, the degree of axonal attrition, and, to some extent, the distance of the lesion from the optic nerve head. Disc swelling is discussed later.
Acquired optic nerve disease is usually heralded by acute or subacute, progressive dimming of central vision. Abrupt onset of monocular visual dysfunction in the age group up to the fifth decade, with a normal-appearing optic disc, is highly suggestive of retrobulbar neuritis, especially if accompanied by dull orbital pain or discomfort of the globe itself. Otherwise, pain is not generally a symptom of optic neuropathies.
In the older age group, the single most common optic nerve disease that presents as apoplectic loss of vision is ischemic infarct of the disc, almost always with disc swelling during the acute phase, for which reason the term anterior ischemic optic neuropathy (AION) has been popularly applied. Retrobulbar disease producing abrupt to subacute loss in the elderly includes cranial arteritis (CA) and meningeal metastases. Slowly progressive monocular visual loss over many months, in neuro-ophthalmologic context, typifies chronic tumoral compression of the optic nerve in its prechiasmal portion.
Insidious bilateral but not necessarily symmetric, central, or cecocentral scotomas are hallmarks of intrinsic optic nerve disease, resulting from nutritional deficiencies, intake of toxins, or heredofamilial atrophies. Rarely, demyelinative disease runs such a slowly progressive course.
When a central field defect is found in one eye, careful search of the temporal field of the contralateral eye is mandatory to rule out the possibility of junctional (nerve and chiasm) compression (Volume 2, Chapter 6, Figs. 1A and C). Most prechiasmal optic neuropathies are caused by inflammatory or vascular disease, whereas practically all chiasmal syndromes are owing to pituitary adenomas, other neoplasms, or aneurysm compression.
Visual loss may be subjectively misinterpreted as sudden under circumstances in which the better eye is momentarily closed or obstructed, or when the involved eye is used in an uncustomary way for monocular viewing (e.g., microscope, telescope, gun sight).
Optic atrophy, with few exceptions, is generally a nonspecific clinical observation, and ophthalmoscopic criteria that permit a retrospective etiologic diagnosis, without other clinical clues, may include arterial attenuation in vascular causes. However, Frisen and Claesson123 quantitatively demonstrated a reduction in central retinal artery caliber of 17% to 24% in nonischemic descending (retrobulbar nerve, chiasm lesions) optic atrophy. Likewise, focal narrowing of retinal vessels correlates best with age and may be seen with a wide variety of optic nerve diseases, including glaucoma.124
Neuroimaging Techniques
With the exception of lesions or fractures of skull bones, MRI has supplanted CT in the elucidation of occult optic nerve disease. By clinical criteria, suspicion may fall on a particular segment of the nerve, such as intraorbital, intracanalicular, or prechiasmal (intracranial), and special anatomic attention should be addressed accordingly. Standard brain studies may provide few sections of orbital or basal skull structures. Therefore, the clinician should provide precise and specific instructions to the radiologist or technician. Relatively thin-section techniques (e.g., 3 mm) are required especially for adequate visualization of the optic canal, paraclinoidal and prechiasmatic portions of optic nerves, with and without gadolinium enhancement. Ideally, high-resolution (1.5–3.0 Tesla unit magnet systems) T1- and T2-weighted views should include axial, coronal, and sagittal sections; oblique views aligned with the long axis of the optic nerve (oblique sagittal) are of dubious value. The fat tissue of the orbit permits excellent contrast because fat appears bright (hyperintense) on T1-weighted images, whereas muscles, vessels, and nerves are darker (hypointense). Moreover, the optic nerve shares MRI characteristics with myelinated white matter of the brain. Blood vessels appear dark because of proton flow voids.125 Protocols that delineate optic nerves from orbital fat and minimize eye movement artifacts, such as fat-suppression fast spin-echo,126 are more effective than conventional T1- or T2-weighted images (Volume 2, Chapter 14, Figs. 19 and 20). Abnormalities of the optic nerve sheath (tumors and inflammation) may be missed if such fat-suppressed and contrasted orbital views are not included. Subsequent illustrations of specific studies follow; a more exhaustive discussion of technical details is beyond the scope of this work, and the reader is referred to available comprehensive reviews.127
Ultrasonography (echography) is a practical, noninvasive adjunct to evaluate anatomic characteristics of optic nerve morphology. Standardized echography is useful in disclosing the presence of perineural fluid, highly suggestive of inflammatory optic neuritis. In addition, perineural fluid accumulation in papilledema may be confirmed and monitored.128
New technologies for imaging the optic disc and quantification of the retinal nerve fiber layer have become commercially available and are now used for clinical and research purposes129 (also discussed later). The most widespread use of such technologies has been for the evaluation of glaucoma but several studies have now targeted nonglaucomatous optic neuropathies as well. For example, such imaging techniques have been used to quantify progression in eyes with papilledema,130 to compare patterns of disc atrophy in arteritic versus nonarteritic anterior ischemic optic neuropathy131 and to measure axonal loss in demyelinating disease.132 Changes in retinal nerve fiber structure have been compared to functional measures including visual field defects,133 full-field visual evoked potential (VEP)134 and multifocal VEP.135 Recent interest has focused on the use of retinal nerve fiber measurement as an indicator of axonal loss in multiple sclerosis, and particularly as a handle on therapies that might offer neuroprotection in this disease.136
THE SWOLLEN DISC: DIFFERENTIAL DIAGNOSIS
Active or passive edematous swelling of the optic disc provides compelling objective evidence of perturbed distal optic nerve function, but by appearance alone it is rarely specific. The causes of optic disc swelling are legion, as outlined in Table 5-7. By definition, the term papilledema is reserved for disc swelling that is caused by increased intracranial pressure. The management of papilledema is different from other causes of acquired disc edema, and therefore this distinction is crucial.
TABLE 7. Etiology of the “Swollen” Disc | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The differentiation of congenitally elevated discs (i.e., pseudopapilledema) from papilledema has already been discussed. In most cases, true papilledema, even in the fully developed form, does not reduce acuity (unless the macula is encroached), nor does it present with field defects other than enlarged blind spots. Therefore, confusion should rarely arise in distinguishing papilledema from inflammatory papillitis or from AION (Table 5-8), two common causes of disc edema that regularly are associated with acute loss of acuity or field and diminished direct light reaction of the ipsilateral pupil. In disc edema accompanying local ocular diseases such as uveitis, central retinal vein occlusion, or postoperative hypotony, there are usually examination findings that point to the correct diagnosis. Simple posterior vitreous detachment137 may rarely be associated with disc and peripapillary hemorrhage and edema. Primary nerve head tumors (melanocytoma, glioma, astrocytic hamartoma, hemangioma) are rare and usually are definable by ophthalmoscopy. Metastatic disc tumors, other than those arising in the adjacent choroid and retina, are extremely infrequent and are characterized by massive hemorrhagic elevation of the disc and peripapillary retina and drastic reduction of vision. Occasionally, the optic nerve head may be infiltrated by leukemia or similar hematologic process, usually with rapidly progressive visual loss.
TABLE 8. Clinical Characteristics of Optic Neuritis, Papilledema, and Ischemic Optic Neuropathy | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
Orbital mass lesions characteristically produce proptosis, but they may also present as chronic, unilateral disc edema with insidiously advancing field loss. As a general rule, unilateral disc swelling should be considered a local vascular or inflammatory disorder of the nerve head or the result of a chronic perioptic mass lesion in the orbit (e.g., nerve sheath meningioma). On rare occasions, papilledema from increased intracranial pressure, including pseudotumor cerebri, may be remarkably asymmetric or even strictly unilateral. When any diagnostic dilemma arises, enhanced CT scan or MRI of brain and orbital optic nerves is mandatory and should be performed sooner rather than later, but obviously after thorough history taking and meticulous examination.
As noted, thin-section (ideally 1.5 mm) contrast-enhanced CT scan or fat-suppressed MRI displays fine details of optic nerve anatomy on both axial and coronal views, and any increase or distortion of nerve diameter is readily detected. Standardized A-scan ultrasonography128 also defines the morphologic characteristics (normal vs. enlarged, solid vs. sheath fluid) of various optic neuropathies; this technique can effectively distinguish among causes of chronic optic neuropathy (perioptic tumor mass vs. remote neuropathy) and disc edema (tumor vs. inflammatory neuritis vs. ischemic neuropathy). Conditions that enlarge the optic nerves include optic neuritis, papilledema of raised CSF pressure, Graves orbitopathy, direct or indirect trauma, perioptic inflammatory pseudotumor, optic glioma, perioptic meningioma, and infrequent infiltrations.
PAPILLEDEMA (RAISED INTRACRANIAL PRESSURE)
The pathogenesis of papilledema (i.e., disc edema owing to increased intracranial pressure [ICP]) has been studied in various acute and chronic experiments. The bulk of evidence indicates that raised pressure is transmitted in the vaginal sheaths of the optic nerves138 with resultant (or attendant?) stagnation of the venous return from the retina and nerve head. That nerve sheath pressure is critical has been demonstrated by Hayreh,139 who showed reversal of disc swelling by opening the nerve sheath. Hayreh139 further established that there is stasis of both fast and slow axoplasmic flow within the optic nerve at the level of the lamina cribrosa. In essence, optic nerve fibers are compressed in the subarachnoid space of the intraorbital portion of the optic nerve owing to elevation of CSF pressure. The subsequent obstipation of intra-axonal fluid mechanics results in swelling of axons and leakage of water, protein, and other axoplasmic contents into the extracellular space of the prelaminar region of the optic disc. This protein-rich fluid adds to the osmotic pressure of the extracellular space of the disc substance. Venous obstruction and dilation, nerve fiber hypoxia, and vascular telangiectasis of the disc are secondary events. Therefore, it is likely that papilledema is primarily a mechanical rather than a vascular phenomenon. It has been suggested140 that the distal retrobulbar portion of the optic nerve sheath (i.e., just behind the globe) is bulbous and distensible and that movement of the globe in the orbit normally milks sheath fluid posteriorly, thus reversing flow and completing the circulation of CSF into the intracranial subarachnoid space. Therefore, perturbation of this pumping mechanism may also play a role in the evolution of papilledema.
The various underlying mechanisms that raise CSF pressure may be summarized as follows: intracranial mass lesions; increased CSF production (e.g., choroid plexus papilloma); decreased CSF absorption; high protein content or cellular debris that obstructs CSF absorption at arachnoidal granulations; obstructive hydrocephalus; increased cerebral blood volume; and increased intracranial venous pressure.141
The clinical picture of chronic unilateral disc swelling most commonly results from obstruction of the subarachnoid space of the ipsilateral nerve by an intraorbital process such as a nerve sheath meningioma (with modest visual loss in the early stage and normal CSF pressure). As noted, on rare occasions, increased CSF pressure, including pseudotumor cerebri syndrome, manifests as truly unilateral papilledema. With respect to intracranial mass lesions, unilateral papilledema has no consistent lateralizing value. Previous optic atrophy may prevent disc swelling of one side, with papilledema developing on the other (the so-called Foster Kennedy syndrome, attributed to subfrontal masses). It is also speculated that a congenital nerve sheath anomaly may obstruct transmission of pressure such that the disc remains flat despite elevated intracranial pressure. However, Muci-Mendoza et al142 demonstrated bilateral optic nerve sheath distention on the CT scan of two patients with pseudotumor cerebri and unilateral papilledema, thus raising the question of mechanisms at the distal portion of the optic nerve. Such anatomic variation may explain the occurrence of increased ICP without disc swelling in either eye, termed idiopathic intracranial hypertension without papilledema or IIHWOP.143,144 The frequency with which this occurs remains controversial.
It usually takes at least 24 hours for the development of papilledema, accounting for its absence in cases of acute elevation of pressure, such as intracranial hemorrhage or craniocerebral trauma.145 Once the intracranial space is decompressed, venous congestion of the disc diminishes rapidly, but disc edema, hemorrhages, and exudates resolve more slowly. Well-developed papilledema resulting from mass lesions takes 6 to 10 weeks to regress after lowering of intracranial pressure.
Early, and even well-developed, papilledema may not be symptomatic. The clinical feature that best differentiates papilledema of intracranial origin from other forms of acquired disc swelling is that visual acuity, field, and pupillary reactions are typically normal, whereas visual function (acuity, field, pupillary reaction) is almost always defective with papillitis (neuritis) or ION. When papilledema has existed for many weeks or months, nerve fiber attrition results in progressive field loss in the form of irregular peripheral contraction and nerve fiber bundle defects. This is the atrophic stage of chronic papilledema, which can ultimately lead to severe visual loss and even blindness. Uncommonly, optic nerve dysfunction is present at initial presentation of a patient with elevated ICP.
Patients with well-developed papilledema may experience brief transient obscurations of vision. Grayouts, blackouts, or other momentary dimming of vision may involve one or both eyes at a time, last seconds, and clear completely. Sudden changes in posture, especially arising, may precipitate such obscurations, or they can occur spontaneously. Such visual disturbance is probably related to transient impairment of nerve head perfusion owing to normal fluctuations of systemic blood pressure particularly associated with postural change. Transient visual loss occurs when the perfusion pressure temporarily fails to overcome the increased resistance in the capillary bed of the distended optic disc. Fluctuations of intracranial pressure may also play a role. Such obscurations are common in the pseudotumor cerebri syndrome. The frequency of obscurations appears to be most closely correlated with high intracranial pressure (at least at the moment of the obscurations) and advanced degree of disc swelling. Because the mechanism of transient obscurations is ischemic, similar to the basis for permanent visual loss in increased ICP, the presence of frequent and/or prolonged episodes should be taken as a worrisome sign of impending visual loss and treatment directed accordingly.
Other signs and symptoms associated with papilledema are related to the underlying pathologic processes that produce the increased intracranial pressure. Headache, nausea and vomiting, and diplopia resulting from lateral recti weakness are typical but nonspecific symptoms of raised CSF pressure, whereas hemiparesis, hemianopias or other field defects, seizures, or specific ocular motility disturbances all have localizing value.
It is helpful ophthalmoscopically to stage papilledema, which on occasion has considerable clinical value. As suggested by Jackson in 1871, papilledema may be classified into four temporal types: early, fully developed, chronic, and atrophic. The early phase of papilledema refers to the incipient disc changes that occur before the development of obvious disc swelling. Blurring of the nerve fiber layer and obscuration of the superior and inferior disc margins are early changes that may actually precede venous engorgement (Fig. 5-26). The use of red-free light (green filter) in the ophthalmoscopic may further delineate early nerve fiber layer changes in incipient papilledema. The veins of the retina ultimately become engorged, tortuous, and dusky, but usually not until disc swelling is well underway.
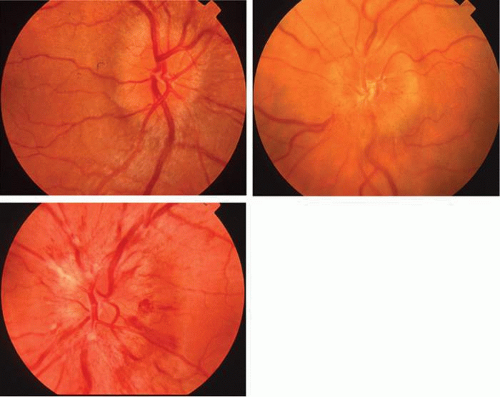 Figure 26. Papilledema. Mild (A), moderate (B), and fully developed (C) disc edema resulting from increased intracranial pressure. Note increasing opacification of the nerve fiber layer, obscuring vessels at the disc surface. |
Spontaneous pulsation of major disc veins is anticipated in approximately 80% of eyes, and its presence militates against the possibility of raised CSF pressure. It is estimated that spontaneous venous pulsation ceases when intracranial pressure exceeds 200 ± 25 mm H2O, but the absence or presence of pulsation is not a consistently useful sign.146 The occurrence of splinter hemorrhages in the nerve fiber layer at or just beyond the disc margin is a major confirmatory finding, especially in the course of repeated fundus observations. However, as noted previously, hemorrhages may infrequently occur with intrapapillary hyaline bodies or in chronic glaucoma, posterior vitreous separation, or for no apparent reason in the elderly.
In early papilledema, as well as in the more fully developed stage, the optic cup is retained. In fact, absence of the central cup is much more likely to be seen in pseudopapilledema than in incipient disc swelling. In the more chronic stage of papilledema, the central cup is likely to be slowly obliterated. As edema progresses, the surface of the disc becomes elevated above the plane of the retina. Nerve fiber layer opacification obscures the scleral disc margins, and minor or major vessels are buried as they course off the disc. At this stage, that is, fully developed papilledema, disc elevation is consistently accompanied by multiple flame hemorrhages, nerve fiber layer infarcts (cotton-wool spots), serpentine tortuosity of veins, and marked disc hyperemia and hypervascularity attributable to telangiectatic dilatation of the superficial capillary bed of the disc surface (Fig. 5-26). Swelling of the nerve fiber layer extends laterally into the retina, so that the area of the nerve head appears enlarged. The retina is raised up from pigment epithelium, and circumferential retinal folds (Paton lines) may be seen around the swollen disc, representing the concentric lateral displacement of retina; these may extend even to the macula (Fig. 5-27A). Rarely, retinal exudates radiate spokelike from the fovea in the form of a star (or half-star between the disc and fovea), with the apex toward the fovea (Fig. 5-27B). If intracranial pressure remains elevated, the acute hemorrhagic and exudative components resolve, and the disc progressively takes on the appearance of the dome of a champagne cork. The central cup remains obliterated, but peripapillary retinal edema resorbs. Small, round glistening hard exudates (axoplasm or protein remnants?) on the surface of the disc may simulate hyaline bodies. This stage of chronic papilledema indicates that disc swelling has been present for months. Nerve fiber attrition is predictable, leading to progressive field loss. As the disc detumesces, pallor slowly emerges, with apparent sheathing of vessels but no real loss of disc substance. Although the disc usually has a milky gray appearance (secondary optic atrophy) (Fig. 5-27C), at times it appears remarkably crisp and white. Even with fairly rapid detumescence of preatrophic papilledema, retinal exudates, changes in the foveal pigment epithelium secondary to edema or subretinal hemorrhage may permanently reduce central acuity.147
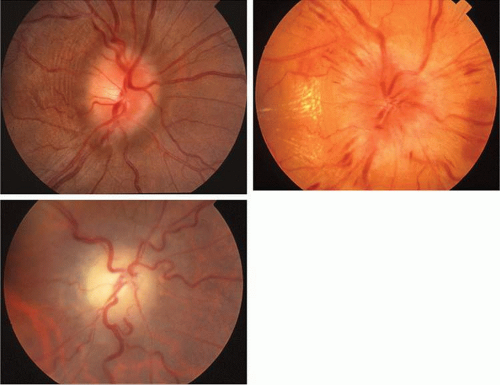 Figure 27. Papilledema. A. Moderate disc edema with Paton lines parallel to the temporal aspect of the disc. B. Marked disc swelling with hard exudates forming a partial star figure tracking to the macula. C. Chronic atrophic papilledema showing fuzzy pallor and loss of nerve fiber layer. |
Choroidal folds, with and without acquired hyperopia, are described in association with papilledema, with neuroimaging that strongly suggests that these striae are likely related to distention of the most distal patulous portion of the optic nerve sheath, which flattens and foreshortens the posterior wall of the globe. It is reported148 that such folds may actually precede disc swelling.
The appearance of the optic discs, sequence of funduscopic changes, and ultimate visual outcome are dependent on variations of intracranial pathology, surgical interventions, and obscure hemodynamic events involving both the disc and the retrobulbar visual pathways. Acute and drastic visual loss may occur, usually owing to ischemic infarction of the disc149 but occasionally secondary to central retinal artery occlusion150 or choroidal neovascularization. In the presence of well-developed, usually chronic, papilledema, cranial decompression, including shunt procedures or ventriculography, is occasionally followed by visual loss that is abrupt or progressive over weeks. Such tragic outcome is neither understood nor predictable, and it may not be prevented even if shunting procedures precede decompression of the primary mass. This event may be related to arterial hypotension provoked by intracranial decompression, especially with the patient in a seated position (e.g., for posterior craniectomy), or to an increase in CSF pressure precipitated by anesthesia. A question of perfusion insufficiency of the distal optic nerve has been raised,151 but the precise mechanism(s) remains elusive.
Although much has been written regarding the use of fluorescein angiography in the diagnosis of papilledema, in instances of ophthalmoscopically definable papilledema, fluorescein fundus angiography is usually superfluous. In eyes in which disc changes are truly debatable, fluorescein angiography may be inconclusive and should not be considered the single indication for further complicated diagnostic studies. Thin-section CT views and fast spin-echo MRI,152 as well as standardized A-scan ultrasonography128 are useful in confirming distended perioptic meninges, and the latter provides a convenient technique to monitor the effects of therapy on nerve sheath size. Optic coherence tomography (OCT) may be used to document and follow changes in optic disc elevation.130
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


