9 The Uveal Tract
NORMAL ANATOMY
THE CILIARY BODY
A precise knowledge of the position of the ciliary body is important in the positioning of surgical incisions for vitreous surgery. The surface markings of the ciliary body from the corneal limbus are 1.5–8 mm on the temporal side and 1.5–7 mm on the nasal side. The anterior third (2 mm) contains the ciliary muscle and ciliary processes, and is known as the pars plicata. The posterior two-thirds—the pars plana—extends posteriorly to the ora serrata where it merges with the retina. There is a dense attachment of the vitreous base over this area and on to the anterior equatorial retina (see Ch. 12).
Overlying the ciliary muscle the epithelium and stroma are thrown up into about 80 ciliary processes. These have a vascular stroma and are covered by two layers of epithelium which are continuous with the iris pigment epithelium anteriorly and with the retinal pigment epithelium and neurosensory retina posteriorly. The inner or superficial epithelial layer is nonpigmented and has tight intercellular junctions. Aqueous is secreted through these cells (see Ch. 7). As in the choroid, the capillaries in the ciliary processes are fenestrated.
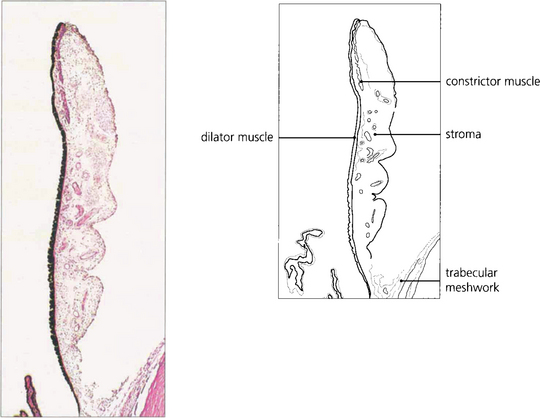
Fig. 9.1 The iris consists of loose, pigmented vascularized tissue anteriorly and a double layer of pigment epithelium posteriorly. The sphincter muscle lies towards the pupillary margin and the dilator muscle in close relation to the pigment epithelium. About 1–2 mm from the pupillary margin on the anterior surface there is a frill known as the collarette. This is the site of the embryological pupillary membrane which atrophies in the eighth month of gestation, and of the minor arterial circle of the iris.
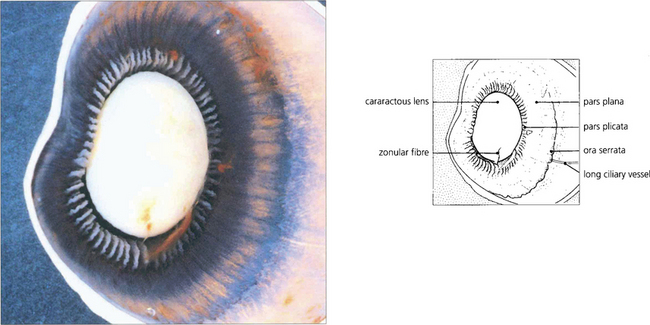
Fig. 9.2 A posterior view of the lens and anterior segment shows the insertion of the retina into the pars plana at the ora serrata and the ciliary processes of the pars plicata. A few remaining zonular fibres can be seen supporting the cataractous lens.
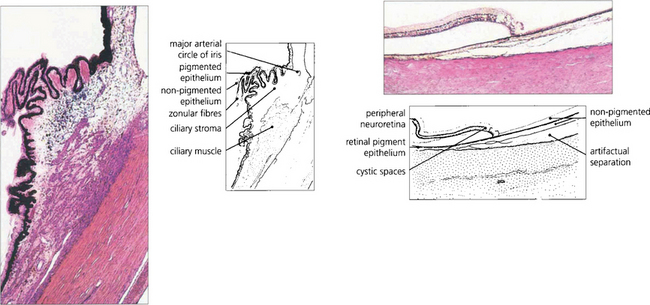
Fig. 9.3 The ciliary body extends from the scleral spur to the ora serrata. Under higher magnification the details of the pars plicata are seen more clearly. Note the two layers of ciliary epithelium, the zonular fibres and the major arterial circle of the iris. At the ora serrata, the neurosensory retina becomes attenuated and cystic, and terminates as the inner non-pigmented epithelium of the ciliary body. The retinal pigment epithelium is continued as the outer pigmented epithelial layer of the pars plana.
BLOOD SUPPLY OF THE UVEAL TRACT
The vascular supply of the uveal tract comes from the posterior ciliary circulation anastomosing anteriorly with the anterior ciliary arteries. The short posterior ciliary arteries leave the ophthalmic artery posteriorly in the orbit (see Ch. 20) and run forwards to penetrate the sclera circumferentially around the optic disc, usually in two major horizontal trunks that divide to supply the optic disc, retrobulbar optic nerve (see Ch. 17) and the choroid. At the disc, two long posterior ciliary branches from these run forward medially and laterally in the lamina suprachoroidia to anastomose with the anterior ciliary arteries adjacent to the major circle of the iris. These long posterior ciliary arteries can frequently be seen in the horizontal meridians of a normal eye if the retinal pigmentation is not too dense. The anterior ciliary arteries are also derived from the ophthalmic artery. They lie on the external ocular muscles (two arteries on the medial, inferior and superior recti, and one on the lateral) and penetrate the sclera at the muscle insertions, and may contribute to the supply of the iris, ciliary body and anterior choroid (although under normal circumstances in a healthy eye the flow is retrograde). The choroidal venous return drains into the orbital veins by the vortex veins, of which there is usually one, but sometimes two, lying in each quadrant of the sclera at the equator.
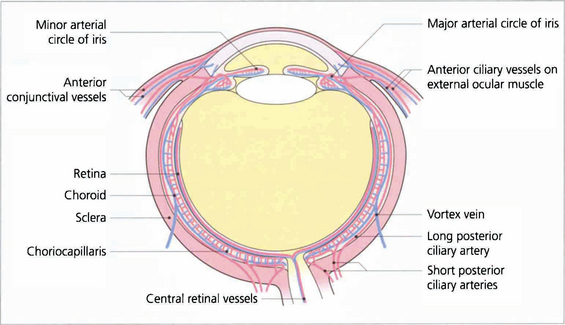
Fig. 9.5 Diagram showing the vascular supply of the choroid by the anterior and posterior ciliary circulations. (Further details of the anterior ciliary and conjunctival circulations are described in Chapter 5 and the optioc disc in Chapter 17.)
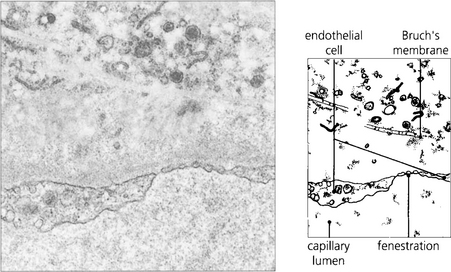
Fig. 9.6 The choroidal arteries divide rapidly to form the choriocapillaris lying beneath Bruch’s membrane. These capillaries have fenestrations between the endothelial cells allowing plasma to leak into the extracellular space. Although there are anatomical anastomoses between the choroidal vessels, physiologically the choriocapillaris functions on a lobular basis.
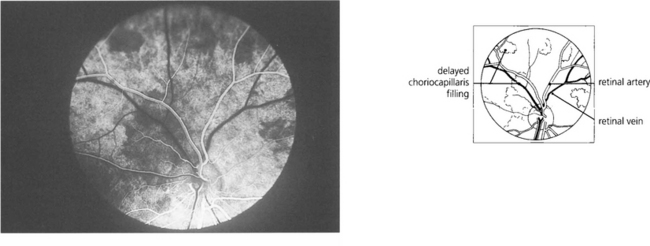
Fig. 9.7 Clinically this is demonstrated in the earliest phases of a fluorescein angiogram as patchy delayed filling of the choroidal bed, and is seen as choroidal infarcts such as Elschnig’s spots or Siegrist streaks (see Ch. 14).
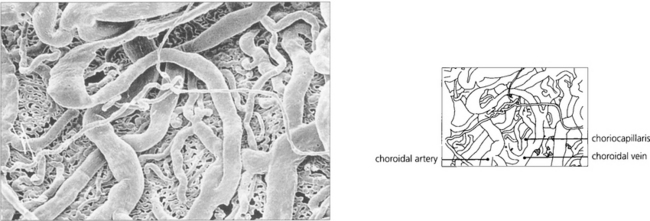
Fig. 9.8 A digest preparation of a cast viewed from the choroidal side in the vicinity of the optic disc shows choroidal arteries supplying the choriocapillaris and drained by the choroidal veins.
By courtesy of Miss J Olver.
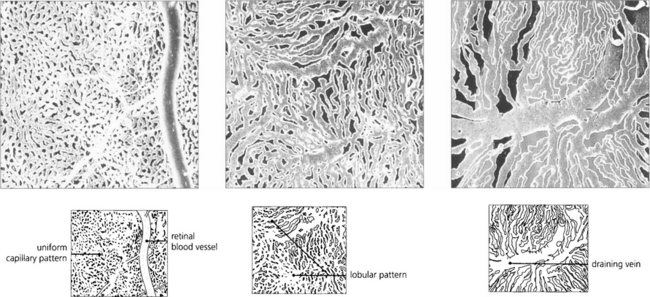
Fig. 9.9 These digest casts of the human choroid show the choriocapillaris from the retinal aspect. At the posterior pole the pattern is uniform (left), in the equatorial fundus a lobular pattern is more apparent (middle), and in the periphery large fan-shaped lobules can be seen (right).
By courtesy of Miss J Olver.
CONGENITAL ANOMALIES OF THE UVEAL TRACT
COLOBOMAS
Colobomas result from defects of closure of the optic cup that occur at 7–8 weeks of fetal life. They can present as a sectorial deficiency varying from the trivial to the gross. They are typically found inferonasally and may involve the iris, choroid and retina, or optic disc (see Ch. 17).
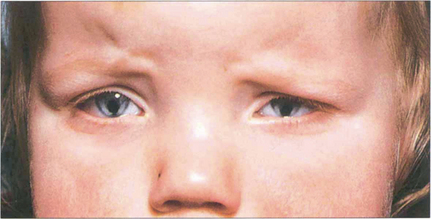
Fig. 9.10 Iris colobomas are sometimes associated with segmental absence of the lens zonules causing a localized indentation of the lens and usually with defects in the choroid and retina. This child with bilateral iris colobomata also has a poorly sighted divergent left eye due to a large chorioretinal coloboma involving the macula.
ANIRIDIA
Aniridia occurs either as a familial autosomal dominant disease or sporadically. The autosomal dominant condition is associated with glaucoma, nystagmus, corneal opacities and photophobia, whereas sporadic cases usually have a high incidence of nephroblastoma (Wilm’s tumour). This is associated with deletion of a tumour suppressor gene, which has been identified on chromosome 11, analogous to the retinoblastoma gene on chromosome 13. All such children require regular screening by renal ultrasonography. A vestigial iris remnant can usually be seen as a frill on gonioscopy (see Ch. 7).
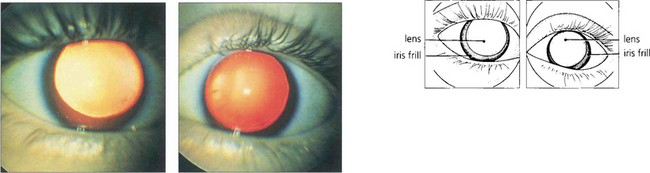
ALBINISM
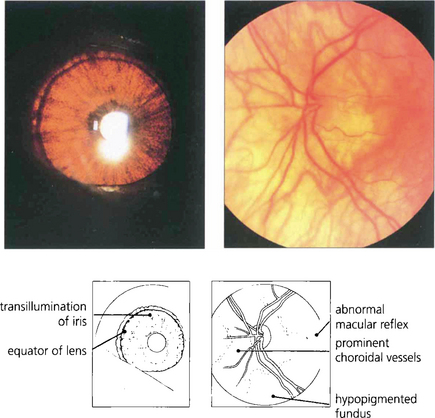
Apart from increased iris transillumination and hypopigmented fundi, albinos with ocular involvement have congenital nystagmus, macular hypoplasia, a high incidence of squint and amblyopia, and an anomaly of the chiasm in which the majority of optic nerve fibres from each eye decussate. This is thought to be caused by the absence of pigmented cells in the chiasm during embryogenesis; these cells ‘direct’ the ingrowing axons. Ocular albinism is a common cause of congenital nystagmus and it is important to examine all such patients for increased iris translucency by iris retroillumination. Excessive pigmentation (melanosis oculi) is discussed in Chapter 3.
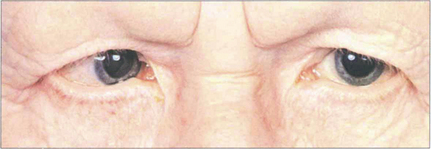
Fig. 9.12 An oculocutaneous tyrosinase-negative patient with congenital nystagmus and a right convergent squint. Note the white eyelashes.
IRIS TUMOURS
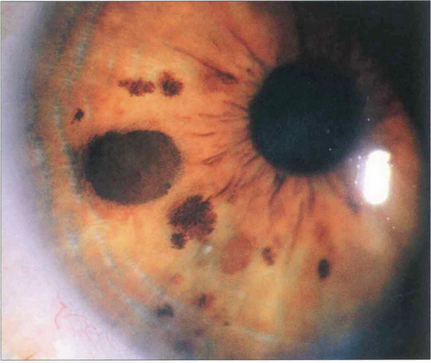
Fig. 9.14 Iris naevi may be pigmented or amelanotic, vascular or avascular and bulky or flat. They may be associated with adjacent ectropion uveae (which is not a sign of malignancy) and can extend into the angle. Iris freckles do not distort normal iris anatomy. Naevi may enlarge or become more deeply pigmented after puberty. Iris naevi can be confused with the so-called iris–naevus (Cogan–Reese) syndrome, a variant of the iridocorneal endothelial (ICE) syndrome (see Chs 6 and 8).
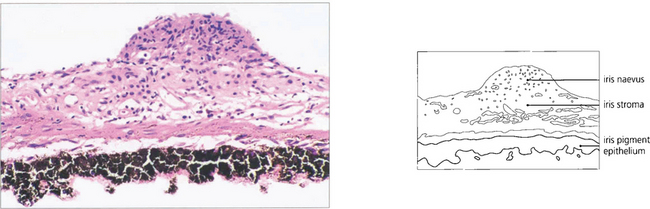
Fig. 9.15 The naevus is formed by proliferation of iris melanocytes which form a nodular layer on the anterior surface of the stroma.
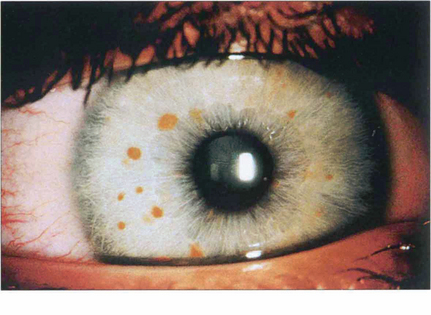
Fig. 9.16 About 90 per cent of patients with neurofibromatosis develop multiple hamartomatous naevi (Lisch nodules) on the stromal surface by their teens. These can occur in both neurofibromatosis types 1 and 2, although they are more common in type 1 (see Chs 2 and 20).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


