The Syndromal Child
Ted L. Tewfik
Congenital anomalies are involved in about half of all pediatric hospital admissions and next to accidents constitute the leading cause of death in children. They involve all medical branches, cross all human medical disciplines, and present worldwide challenges. The significance of these birth defects and the accurate diagnosis of each condition or of complex syndromes are often difficult to determine.
The goal of any dysmorphic assessment is to interpret the pattern of structural anomalies correctly and arrive at the diagnosis. The practicing specialist dealing with the ear, nose, and buccopharyngeal region is confronted frequently with problems related to congenital anomalies. The spectacular advances in basic and clinical genetics during the past three decades have brought congenital malformations and inherited disorders to the forefront of medical attention and care. Congenital anomalies affect 3% of neonates; by age 5 years, as more subtle anomalies manifest, the percentage climbs from 7% to 10%. The shared spectra of dysmorphic phenotypes and their inherent variability can be overwhelming. Several thousand distinct syndromic entities have been described, and because of their rarity, the typical specialist will not encounter the vast majority of them. The purpose of this chapter is to provide the otolaryngologist with a systematic approach to the dysmorphic neonate, infant, or child and to demonstrate that dysmorphology is a learned discipline that can be practiced by any physician who is inquisitive, thorough, and willing to cultivate his or her powers of observation.
Inheritance of traits and disorders may be monogenic or multigenic or the result of the transmission of an abnormal chromosome (e.g., a deletion or a duplication). The expression of specific genes is influenced by other genes or environmental conditions. Besides the classical mendelian patterns of inheritance, several nonclassical patterns of inheritance have been discovered and studied in recent years.
Single genes are usually inherited according to one of the following mendelian patterns: autosomal dominant (AD), autosomal recessive, X-linked dominant, and X-linked recessive. These patterns may be apparent in pedigrees, the proper drawing of which is the first step in the study of inherited diseases in families. They should be included in all case histories in which a familial disease is suspected. They are also essential for linkage studies, gene mapping, genetic counseling, and prenatal diagnosis. The symbols commonly used are standard and generally accepted (Figs. 107.1, 107.2, 107.3).
A person is homozygous for a certain gene when both alleles at the same locus are identical. Heterozygosity implies the presence of two different alleles. Usually the term is used for situations when one of the alleles bears a pathogenic mutation.
There is no absolute distinction between “dominant” and “recessive” genes. By definition, a “recessive” gene has no detectable expression in a heterozygote under the conditions of study and analysis. Diseases resulting from deficiency of enzymatic activity behave as recessives because, in the heterozygous situation, the amount of enzymatic activity produced by the unaffected allele allows a normal metabolic function. When the gene codes for an abnormal structural protein, the mutant allele usually acts as a dominant. In that case, the cells of the heterozygote carry the mutant gene and synthesize a mixture of the structurally normal and abnormal proteins. As a result of changes in the physical properties of the protein, heterozygotes may show phenotypic abnormalities (1, 2).
The following are the characteristics of AD inheritance:
At least one of the parents of an affected person is also affected, except when the penetrance is reduced or the trait arises by a new mutation or when there is germline mosaicism. The trait appears in every generation. There is no “skipping” except when the “penetrance” is reduced.
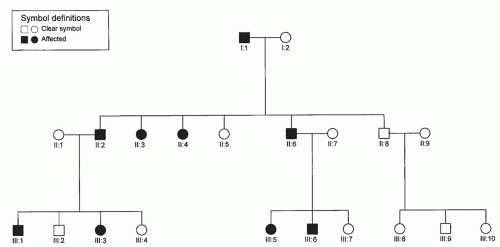
Figure 107.1 Autosomal dominant inheritance. Vertical pattern of trait expression; males and females are equally affected.
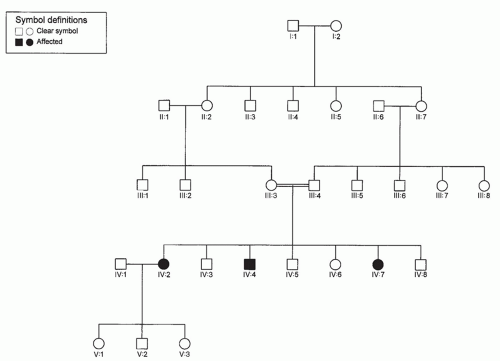
Figure 107.2 Autosomal recessive inheritance. Horizontal pattern of trait expression. Parental consanguinity is often present in families with autosomal recessive disorders.
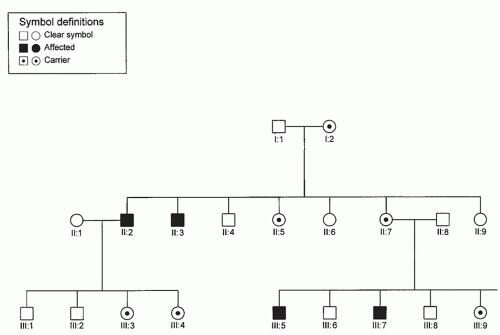
Figure 107.3 X-linked recessive inheritance. Males only are affected. There is no male-to-male transmission, and daughters of affected males are carriers.
An affected person heterozygous for the trait has an equal chance of transmitting the mutant or the normal allele to each child.
Males and females can have and transmit the trait equally.
Two traits are said to be codominant when their genes are allelic and are both expressed in a heterozygote individual. For example, in a person with blood group AB, genes for both groups A and B are expressed.
The following are the characteristics of autosomal recessive inheritance:
The parents are heterozygous and phenotypically normal; the trait appears only in the children who are homozygous for the mutant gene.
Statistically, one-fourth of the children of heterozygous parents are affected and thus there is a chance of 25% at each pregnancy.
Males and females have equal chance of being affected.
When an individual carries two different mutated alleles at the same locus, the condition is called a genetic compound. In consanguineous marriages, both partners have a higher risk of being carriers of the same rare recessive gene. Thus, the children who are products of consanguineous marriages run an increased risk of being affected with autosomal recessively inherited conditions. This risk may be increased by 100-fold. However, in absolute terms, it is of the order of 1%.
X-linked recessive inheritance occurs when the mutant gene is on the X chromosome. It has the following characteristics:
The trait is much more common in males than in females. Females have two X chromosomes, and males have only one. All males carrying the X-linked gene express the trait. Females are affected when they are homozygous and in certain rare heterozygous cases, as an effect of the Lyon hypothesis, when a high percentage of cells have random inactivation of the X chromosome carrying the normal allele.
An affected man passes his X-linked gene to all of his daughters who thus become carriers but are not affected. He does not transmit it to any of his sons.
A carrier woman passes her X-linked gene to half of her sons who manifest the trait and to half of her daughters who become carriers.
X-linked dominant inheritance has the following characteristics:
Affected men transmit the trait to all their daughters who then display the clinical manifestations of that particular condition. They transmit the gene to none of their sons.
Heterozygous women are affected. They transmit the trait to half of their sons and half of their daughters.
Y-linked genes occur only in males. They are transmitted to all the sons and none of the daughters. There are very few genes known to be definitely on the Y chromosome.
PENETRANCE
Certain traits may be modified by other genes and by environmental factors to such an extent that they may not be recognizable either clinically or by laboratory tests, although the gene causing the trait is present. In such cases, the gene or trait is said to have incomplete or reduced penetrance. Thus, certain AD traits with reduced penetrance may “skip” a generation.
EXPRESSIVITY
Expressivity indicates the variation in expression of a phenotype that a gene may have. In clinical medicine, a certain gene with variable expressivity may produce mild, moderate, or severe disease. A forme fruste is an extremely mild and clinically insignificant expression of a condition. Variable expressivity and formes frustes are noted especially in AD inheritance. For example, certain patients with cleidocranial dysostosis may not be identified clinically and are diagnosed in retrospect by special radiographic testing when other more affected members of their families are detected.
PLEIOTROPY
Pleiotropy is the occurrence of multiple phenotypic effects of a mutant gene or gene pair. For example, the primary defect in Hurler syndrome is the specific deficiency of α-L-iduronidase, with accumulation of the substrate in the lysosome. Secondary effects are mental retardation, craniofacial anomalies, dysostosis multiplex, corneal opacification, and hepatosplenomegaly.
HETEROGENEITY
A condition is causally heterogeneous when multiple causes result in the same effect. Genetic heterogeneity of a disease may result in confusing pedigrees. Thus, inherited congenital neurosensory deafness in different families may be the result of two autosomal recessive genes at two different loci. The children of two deaf individuals, each homozygous for a recessive trait at a different locus, may all have normal hearing because they will be heterozygous for two separate loci.
The craniosynostoses are also genetically heterogeneous. Thus, Pfeiffer syndrome may be the result of mutations of FGFR1 (fibroblast growth factor receptor 1) on chromosome 8 (8p11.2-p12) or FGFR2 on chromosome 10 (10q25.3-q26).
Moreover, many malformations are pathogenetically heterogeneous, separate mechanisms being responsible for the same effect. Robin sequence is an example. It is commonly present in Stickler syndrome, which is a singlegene, AD condition. Alternatively, it may be the result of predominantly environmental conditions in utero, such as oligohydramnios, in which case the chin is compressed against the sternum, thus restricting mandibular growth and impacting the tongue between the palatal shelves (3).
MALFORMATION
A malformation by definition is a structural morphologic defect of an organ, part of an organ, or a larger region of the body resulting from a developmental process that is intrinsically abnormal.
A major malformation is one that requires intervention, represents a significant medical or psychologic challenge, and may seriously impair or prevent normal functioning. Examples are ambiguous genitalia and anencephaly. A minor malformation is one that does not represent a lifelong medical or psychologic challenge. Such anomalies can be corrected (unilateral cleft lip, postaxial polydactyly). Minor malformations and variants are important flags for the dysmorphologist. One or two may be found in otherwise normal individuals, but the simultaneous finding of three or more minor anomalies has special significance. In such a patient, the clinician should be prompted to search for occult major malformations and consider a syndromic diagnosis.
Some hereditary traits are transmitted in a nonmendelian pattern, and they cannot be classified as AD, recessive, or X-linked. Mosaicism, uniparental disomy, genomic imprinting, and mitochondrial inheritance are examples that are not discussed here.
SYNDROME
The term syndrome is used to describe a broader error of morphogenesis in which the simultaneous presence of more than one malformation is known or assumed to be the result of a single etiology.
DEFORMATION
Abnormalities in form, shape, or position resulting from normal responses of the affected tissues to the presence of nondisruptive mechanical forces are known as deformations. Potter sequence is an example of deformation.
DISRUPTION
Disruption is when an originally normal developmental sequence is the victim of a process causing ischemia, tissue breakdown, or both.
The most commonly observed disruption is the result of early amnion rupture when entanglement of the fetus in free-floating amniotic bands produces partial or complete amputation of normally formed structures such as digits, limbs, or bizarre facial clefts (3).
SEQUENCE
The term sequence is used to designate a series of anomalies resulting from a cascade of events initiated by a single malformation, deformation, or disruption.
ASSOCIATION
An association is the nonrandom occurrence in two or more individuals of multiple anomalies not known to represent a polytopic field defect, sequence, or syndrome.
A PRACTICAL APPROACH TO THE INFANT OR CHILD WHO IS SYNDROMIC
Given the fact that several thousand distinct syndromic entities have been described, one cannot possibly successfully use a vague memorization strategy in dysmorphology. Instead, what is necessary is a systematic approach that concentrates on meticulous attention to the details of history and physical examination, combined with a method that allows the physician to sift through these details and come up with the best “clues.”
History
The elements of the dysmorphologic history are identical to those of a general pediatric history, but the physician must be particularly tenacious in obtaining specific details. Keep in mind that the parents of a newborn with congenital anomalies may be unable to provide a complete and coherent account of events during the pregnancy and labor and may not have been fully aware of the nature or extent of abnormal ultrasound findings.
If you are assessing an older child, the parents may not recall many obstetric details or they may inadvertently overemphasize the importance of particular prenatal or peripartum events in an effort to explain their child’s difficulties. When possible, it is always best to obtain the relevant medical records.
Physical Examination
The dysmorphology examination differs significantly from a general pediatric examination in its emphasis and attention to detail. The sequence of the examination varies with the examiner, but it should be systematic to prevent omissions.
Precise measurements with comparisons made to standardized curves are helpful in defining the problem. The most useful clues are often the most subtle. A single minor anomaly can be found in up to 30% of newborns. However, less than 10% of neonates have two or more minor anomalies, and only 4% have three or more. The presence of three unrelated minor anomalies is associated with a 20% risk of major structural defects, and most of these infants will, in fact, have a dysmorphic syndrome.
It may be necessary to examine family members, particularly for the presence of minor variants detected in the proband. Examination of the family members may also help you decide whether a mildly dysmorphic facial feature is really only a familial trait. Most parents will be happy to provide you with access to the family photo album, from which age-appropriate comparisons with your patient can be made.
Interpreting the Findings
Having identified the pertinent elements of your patient’s history and physical examination, the next step is to interpret your findings from a developmental and morphologic viewpoint.
Genetic counseling is based on averaged data pooled from the general population. Information should be presented to the parents ensuring that they understand the different issues related to recurrent risks, inheritance, and prognosis.
In summary, the spectrum of dysmorphic phenotypes is broad and includes malformations, deformations, disruptions, sequences, associations, and syndromes. An effective assessment involves a meticulous history and physical examination combined with a developmental and morphologic interpretation of the findings. Additional clues are provided by ancillary investigations and the use of computer databases and literature searches. In attempting to serve the patient and family, our efforts to identify, categorize, and rationalize the abnormal will continue to provide exciting revelations into normal processes of human development. Ultimately our understanding of these processes will be greatly enhanced by rapidly advancing deoxyribonucleic acid technology. A valuable tool accessible on the World Wide Web is Online Mendelian Inheritance in Man (www3.ncbi.nlm.nih.gov/omim/). Other databases include the London Dysmorphology Database and POSSUM.
Many classifications for the congenital syndromes have been attempted; we found that the following are more inclusive. We present two classifications: one based on system involvement and the other based on etiology. A few examples are included under the different headings.
I. Classification of Otolaryngologic Syndromes and Conditions (based on system involvement)
Syndromes associated with ocular disease Usher, Norrie, Fraser, Alstrom, Bardet-Biedl
Syndromes associated with craniofacial anomalies
Craniosynostosis
Apert, Crouzon, Pfeiffer, Saethre-Chotzen, Jackson-Weiss, Carpenter, Antley-Bixler
Abnormal contour
Encephalocele (with absent corpus callosum, clefting, Dandy-Walker and Arnold-Chiari malformations, ectrodactyly, hypothalamic pituitary dysfunction)
Orofacial clefting
Facial clefts and associated anomalies, Tessier clefting system, lateral facial clefts, oblique facial clefts, median mandibular defects
Branchial arches
Goldenhar, Treacher Collins, Nager, Miller, Wildervanck, Möbius, oro-facial-digital syndromes (I-VIII)
Unusual facies
Opitz BBB/G, Noonan, Robinow, Binder, Coffin-Siris syndromes
Syndromes associated with nervous system disease Neurofibromatoses (1, 2, 3, 4, 5, 6, 7, 8), Melkersson-Rosenthal, Cockayne, Friedreich ataxia, Hallgren, Prader-Willi
Syndromes associated with integumentary system disease Waardenburg, Leopard
Syndromes associated with musculoskeletal anomalies ichthyosis syndromes (Desmons, KID), oculocutaneous albinism syndromes, Ehler-Danlos syndromes
With contractures
Multiple pterygium, Whistling face, Schwartz-Jampel, Marden-Walker
Abnormal stature
Short
Aarskog, Cornelia de Lange, Hallerman-Streif, Rubinstein-Taybi, Silver-Russell, Bloom, Seckel
Moderate
Smith-Lemli-Opitz
Overgrowth
Sotos, Marshall-Smith, Weaver, Beckwith-Wiedeman
Senile appearance
Progeria
Osteochondrodysplasia
Achondrogenesis (types I-IV), achondroplasia, campomelic dysplasia, chondrodysplasia punctata, Kniest, Nance-Sweeney, osteogenesis imperfecta (OI) (types I-IV)
Other skeletal disorders
Craniometaphyseal dysplasia, Van Buchem disease, sclerosteosis, Camurati-Engelmann disease, cleidocranial dysplasia, Marfan syndrome, McCune-Albright, Stickler
Syndromes with urogenital anomalies
Branchio-oto-renal (BOR), Alport, Renal tubular acidosis
Syndromes with metabolic (and endocrine) anomalies Mucopolysaccharidoses (I-VII), mannosidosis, gangliosidoses, lipodoses, diabetes and associated syndromes, Pendred, Kallmann, Perrault
Hamartoneoplastic syndromes
Sturge-Webber, Von Hippel-Landau, Goltz, Peutz-Jegher, Maffuci
Gingivodental syndromes
Gingival fibromatosis and its syndromes, Rieger, tricho-dento-osseous, LADD
Syndromes associated with environmental and teratogenic agents*
Fetal alcohol, rubella, thalidomide, vitamin A
Chromosomal syndromes*
Trisomy 21, trisomy 13, trisomy 18, cri-du-chat, Wolf-Hirshhorn, Turner, FragileX
Associations*
CHARGE, MURCS, VATER, or VACTERL
Sequences*
DiGeorge, Klippel-Feil, Robin, Potter
Spectra*
Facio-auriculo-vertebral, oromandibular limb hypogenesis
Miscellaneous syndromes*
Jervell and Lange-Nielsen, Kartagener, Shprintzen, Coffin-Lowry, Kabuki make-up, Keutel, Pallister-Hall, Floating-Harbor
II. Classification of Otolaryngologic Syndromes and Conditions (Based on Eetiology)
Chromosomal abnormalities
Turner, Down (trisomy 21), trisomy 13, trisomy 18, cri-du-chat, 4p-, 9p-
Microdeletion chromosomes
Velocardiofacial (Shprintzen), Williams, Rubinstein-Taybi, Langer-Gideon
Single gene disorder
AD: achondroplasia, Marfan, neurofibromatosis (NF), Noonan, Apert, Crouzon
Autosomal recessive: phenylketonuria (PKU), Bardet-Biedl, Smith Lemli Opitz
X linked: Aaskog, ichthyosis, aqueductal stenosis
Mitochondrial
Environmental and teratogenic
Rubella, thalidomide, dilantin, PKU embryopathy
Multifactorial
Anencephaly, cleft lip/palate, congenital heart disease
Syndromes with uncertain etiology
Klippel-Feil, Cornelia-de Lange, associations
APERT SYNDROME
Acrocephalosyndactyly Type I
The major diagnostic criteria are the craniosynostosis and syndactyly of the hands and feet. The skull is acrocephalic with a shortened anteroposterior diameter and a flat occiput. The craniosynostosis involves the coronal suture. However, the acrocephalic feature of Apert syndrome is not only due to synostosis of the coronal suture, but is a result of the primary disturbance in growth of the cranial base with precocious endochondral ossification (4). The forehead is prominent. The fontanelles are large and close at a later age. The palpebral fissures are downslanting. The corners of the mouth are downturned, and it is sometimes described as “trapezoid mouth.” Dental anomalies include delayed and ectopic
eruptions and shovel-shaped incisors. Malocclusion with mesial molar occlusion and severe crowding of the teeth are also frequently noted. A significant number of patients with Apert syndrome are mentally retarded. Malformations of the corpus callosum, the limbic structures, or both do occur. Other findings include gyral abnormalities, megalencephaly, hypoplasia of the white matter, and heterotropic gray matter. Of patients, 70% have normal intelligence (4, 5).
eruptions and shovel-shaped incisors. Malocclusion with mesial molar occlusion and severe crowding of the teeth are also frequently noted. A significant number of patients with Apert syndrome are mentally retarded. Malformations of the corpus callosum, the limbic structures, or both do occur. Other findings include gyral abnormalities, megalencephaly, hypoplasia of the white matter, and heterotropic gray matter. Of patients, 70% have normal intelligence (4, 5).
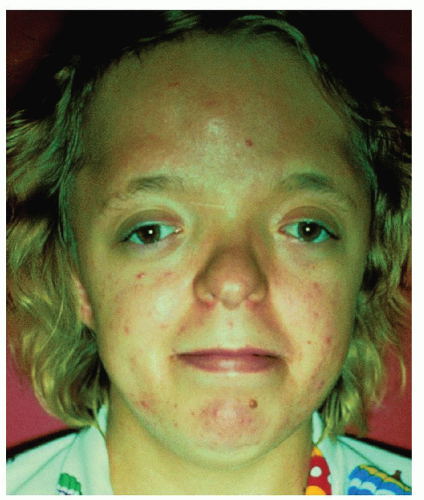 Figure 107.4 Apert syndrome: Typical facies of Apert’s syndrome. Note antimongoloid slanting of the palpebral fissures, exophthalmos, hypertelorism, oxycephaly, and midfacial hypoplasia. Photo Courtesy of Meinhard Robinow, MD from Gold DH, Weingeist TA. Color atlas of the eye in systemic disease. Baltimore, MD: Lippincott Williams & Wilkins, 2001, with permission. |
The pituitary fossa and the basiocciput are significantly larger than normal in patients with Apert syndrome. Anomalies of the hands include syndactyly of the second, third, and fourth fingers that form a mid-digital mass (Fig. 107.4). The first and fifth fingers may join the hand mass or be separate from it. The distal phalanx of the thumb is often broad. Syndactyly is also noted in the feet and involves the second, third, and fourth toes. Fusion of the tarsal bones and an extra metatarsal have been noted. Other anomalies include abnormalities of the cervical vertebrae (single fusion 37%, multiple fusion 31%). C5 and C6 are often involved. Moderate to severe acne at adolescence occurs in 70% of patients. Synostosis of the radioulnar complex, pyloric stenosis, ventricular septal defects, and polycystic kidneys are rare occurrences.
The nasal bridge is depressed, and the midface usually is hypoplastic. The palate is narrow and is described as “byzantine-arch shaped.” Cleft palate may also occur (30%). Anomalies of the ears include otitis media, conductive hearing loss, fixation of the stapedial foot plate, and wide cochlear aqueduct. Temporal bone studies revealed absence of the internal auditory canals (IACs) in one patient and enlarged subarcuate fossa in another report. Anomalies of the airway as well as tracheal stenosis, obstructive sleep apnea (OSA), and cor pulmonale with resultant early death were also reported (3, 4). In general, OSA is related with a lower quality of life in children with syndromic craniosynostosis. Behavioral problems are more common in boys with Apert syndrome. OSA-18 scores are correlated with OSA severity (4).
The syndrome is AD inherited. An increased paternal age is a factor in sporadic cases, which accounts for the majority. The incidence of Apert syndrome is estimated to be 15.5 per 1 million, and it represents 4.5% of all cases of craniosynostosis. Mutations have been found in FGFR2 (3). The primary site of the acrocephalic feature in Apert syndrome is a dwarf cranial base with accelerated chondrocytic differentiation due to aberrant activation of the FGFR2 signaling (5).
BRANCHIOOTORENAL SYNDROME
Melnick-Fraser Syndrome
The association of auricular malformations, branchial fistulae, deafness, and renal anomalies was first described in 1975. The prevalence is estimated to be 1:40,000 in the general population.
Renal dysplasia is reported in more than two-thirds of patients and varies from tapered superior poles (duplication of the collection system) to marked agenesis of the kidney. The syndrome may include Potter sequence. Mitral valve prolapse is a new finding in BOR syndrome. It was reported in five individuals from the same family (1, 7).
The preauricular pits are blind, pin head-sized depressions in the superior part of the pinna of the ear. They are seen in 75% to 85% of the patients. Other external ear malformations (40% to 60%) may include preauricular tags, lop or bat ears, and microtia (Fig. 107.5A and B). The external auditory canals (EACs) may be atretic. Anomalies of the middle ear include anomalies of the ossicles, facial nerve, and fallopian canals. Computed tomography (CT) of the temporal bones may demonstrate hypoplastic cochlea and absent or hypoplastic semicircular canals. Mondini dysplasia, widened vestibular aqueduct, and vestibular sac were also reported. The hearing loss is usually stable and may be conductive, sensorineural, or mixed. It is estimated that 2% of profoundly deaf children have BOR syndrome. The branchial fistulae (63%) are usually bilateral in the lower part of the neck, and the external openings are on the medial border of the sternomastoid muscle. Other associated manifestations include aplasia or stenosis of the lacrimal ducts (8% to 9%), high-arched or cleft palate, deep overbite, and facial nerve anomalies.
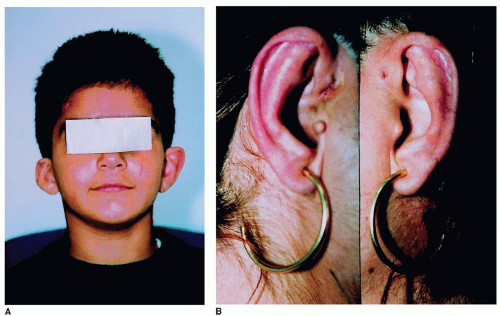 Figure 107.5 A,B: Branchiootorenal syndrome (BOR) syndrome. Note the ear deformity in (A) and the preauricular pit in (B). From Pierides AM, Athanasiou Y, Demetriou K, Koptides M, Deltas CC. A family with the branchio-oto-renal syndrome: clinical and genetic correlations. Nephrol Dial Transplant 2002;17:1014-1018. |
BOR syndrome is AD with high penetrance and variable expression. Pathogenesis is presumed to be the result of deficiency in the differentiation of the first and second branchial arches. The anomalies of the renal system are the result of abnormal interaction between the ureteric bud and the metanephric blastema. It is interesting to note that the inner ear (stria vascularis) and the renal glomeruli share a common antigen and more than one group has described histopathologic changes on temporal bone examinations (1). Mutations in EYA1 are the most common cause of branchiootic syndrome. Large chromosomal aberrations of 8q13, including complex rearrangements, occur in about 20% of these individuals. However, submicroscopic deletions and the molecular characterization of genomic rearrangements involving the EYA1 gene have been rarely reported (1, 2, 6, 7).
CHARGE ASSOCIATION
The components of the CHARGE condition are as follows: C (coloboma of the eye), H (heart disease), A (atresia of the choanae), R (retarded development and growth), G (genital anomalies), and E (ear anomalies, deafness, or both).
Choanal atresia is associated with more than 65% of cases. It is bilateral in more than two-thirds of the patients. In unilateral cases, it is more common on the left side. The colobomatous malformations are seen in 80% of the cases. They range from iris coloboma with intact vision to anophthalmia and retinal colobomas. Microphthalmia and nystagmus may also occur. Cardiac anomalies may include tetralogy of Fallot, patent ductus arteriosus, ventral septic defects, atrial septic defects, coarctation of the aorta, or rightsided aortic arch. Mental deficiency is reported in the majority of cases, ranging from mild to severe. Central nervous system (CNS) malformations have also been described, such as arrhinencephaly or holoprosencephaly. Agenesis of the internal carotid artery was recently reported (1, 8).
Minor kidney anomalies, cryptorchidism, microphallus, congenital hypothyroidism, imperforate anus, and pectus carinatum have also been reported.
Anomalies of the external ear include low-set ear or posteriorly angulated, asymmetric, or cup-shaped pinnae (Fig. 107.6). Anomalies of the inner ear have been coined as “charge dysplasia of the temporal bone”—which includes Mondini dysplasia of the pars inferior and complete absence of the pars superior. The hearing loss is conductive or sensorineural and often asymmetric (1).
Other anomalies include Robin or DiGeorge sequence, facial asymmetry, micrognathia, feeding difficulties,
tracheoesophageal (TE) fistula, esophageal atresia, cleft lip or palate, and upper airway abnormalities.
tracheoesophageal (TE) fistula, esophageal atresia, cleft lip or palate, and upper airway abnormalities.
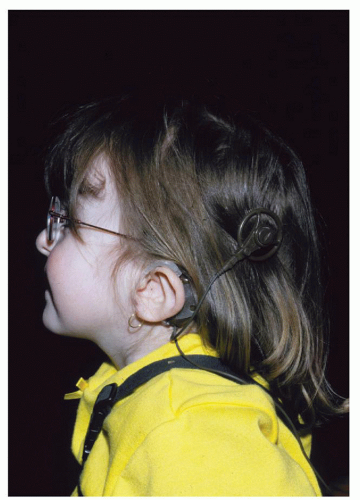 Figure 107.6 A CHARGE association. Note the low-set posteriorly angulated pinna. Description = CHARGE syndrome: unusually shaped ears showing cochlear transplant. From Blake KD, Prasad C. CHARGE syndrome. Orphanet J Rare Dis 2006;1:34. |
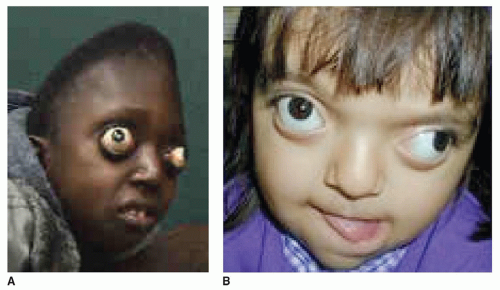 Figure 107.7 A,B: Patients with Crouzon syndrome. Note the proptosis in both patients due to the shallow orbits. A: From http://www.mymultiplesclerosis.co.uk/misc/petero-byakatonda.html, with permission, B: Courtesy of Dr. Stephen R. Cohen, with permission. |
In most cases, the syndrome is sporadic. The CHARGE association is most probably the result of an abnormality in the migration or development of the cephalic neural crest cells (1, 2). AD or recessive inheritance has been suggested. Translocations involving chromosome 6 and 8 have been reported. Other chromosomal abnormalities have also been suggested. The CHD7 gene on chromosome 8q12.1 was recently shown to be a major gene involved in the etiology of CHARGE syndrome (2, 8).
CROUZON SYNDROME
Craniofacial Dysostosis
Craniofacial dysostosis is characterized by cranial deformity resulting from premature craniosynostosis, hypoplastic midface, exophthalmos, hypertelorism, and mandibular prognathism. The syndrome is estimated to occur in 16.5 people per 1 million.
Craniosynostosis of the coronal, sagittal, and lambdoid sutures may occur, giving the skull a brachycephalic shape. Occasionally, scaphocephaly or clover leaf deformity may occur. Crouzon syndrome constitutes approximately 4.8% of cases with craniosynostosis in general. Proptosis occurs in most cases secondary to the shallow orbits (Fig. 107.7A and B). It may result in exposure keratitis. Optic nerve involvement is usually evident in half of the cases. Other ocular findings include megalocornea, nystagmus, keratoconus,
ectopia lentis, or colobomas of the iris. Frequent headaches, seizures, and mental deficiency may also occur. Acanthosis nigricans has been described in association with Crouzon syndrome. The surgical treatment is usually directed toward correcting craniosynostosis. The facial abnormalities may be corrected using the Tessier procedure.
ectopia lentis, or colobomas of the iris. Frequent headaches, seizures, and mental deficiency may also occur. Acanthosis nigricans has been described in association with Crouzon syndrome. The surgical treatment is usually directed toward correcting craniosynostosis. The facial abnormalities may be corrected using the Tessier procedure.
It is estimated that 30% to 55% of patients with Crouzon syndrome have hearing loss. The hearing loss is usually conductive. The EACs may be atretic. Ossicular fixation and deformities have been described as well. Anomalies of the palate include lateral palatal swellings in 50% of cases, but cleft lip or palate is not common. The maxillary teeth are often crowded, and there is usually an anterior open bite. The nose is often parrotlike. Calcification of the stylohyoid ligament occurs most of the time. Anomalies of the cervical vertebrae involve mostly C2 and C3.
Surgical intervention involves fronto-orbital and midfacial advancement, usually done in stages in childhood. The occlusion is corrected adequately with orthodontics and final maxillary advancement after skeletal growth is completed (1).
DOWN SYNDROME
Trisomy 21 or Down syndrome is the most common of the chromosomal abnormality syndromes. The diagnosis of Down syndrome can be made in the early newborn period. Four out of 10 signs are needed to make a clinical diagnosis (Table 107.1). However, a final diagnosis may be made only after having the results of the chromosomal pattern. Adaptive behavior in Down syndrome is described to increase until middle childhood and to begin to decline in adolescence, whereas significant deterioration in middle adulthood has been attributed to early onset of dementia. Nevertheless, opinions diverge about when the slowing down of adaptive and cognitive abilities starts. In a recent prospective cross-sectional study, Dressler et al. concluded that individuals with Down syndrome continue to increase competence in adaptive behavior until age 30, even when cognitive abilities reach a plateau. There was no major decline in middle adulthood. This may be due to exposure to daily life, but needs to be supported by further studies.
TABLE 107.1 TEN CARDINAL FEATURES OF DOWN SYNDROME IN THE NEWBORN | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The eyes have speckled irises (Brushfield spots), and fine opacities are seen in the lens on slit lamp examination. Mental deficiency is a hallmark of the syndrome. The intelligence quotient (IQ) varies between 30 and 50. Various brain lesions have been described including abnormalities of the cerebellar peduncles and fibrous gliosis of the white matter. The frequency of epilepsy is estimated to be 5%. Older patients develop changes characteristic of Alzheimer disease. Cardiac anomalies are seen in approximately 40% of patients; atrioventricular communis or ventricular septal defects, Fallot tetralogy and patent ductus arteriosus occur in decreasing order of frequency. Atlantoaxial instability is seen in 10% to 20%. The hands have short metacarpals and phalanges with clinodactyly and hypoplastic midphalanx of the fifth finger. Single palmar creases and ulnar loop dermal ridge patterns are seen in a high percentage of cases. There is a wide gap between the first and second toes. Male patients have a small penis and low testosterone production and are almost all infertile. The hair is often sparse; cutis marmorata and dry hyperkeratotic skin are not uncommon (1).
Other uncommon anomalies include TE fistula, incomplete fusion of the vertebrae, cryptorchidism, syndactyly, and keratoconus. The main cause of death is the cardiovascular malformation and is highest in the first 2 years of life. Leukemia occurs in 1% of patients. Lymphomas, testicular carcinoma, and CNS tumors have also been reported. Flat facial features, small or absent paranasal sinuses, hypertelorism, small nasal bridge, and oblique palpebral (upward-slanting) fissures occur in more than 80% of patients. The tongue is relatively large and protrudes from the mouth. It may be furrowed or fissured. Occasionally true macroglossia exists. The ears are small with overfolding of the superior part of the helix. The EACs are stenotic, especially at the isthmus (bony-cartilaginous junction). Conductive hearing loss occurs in 60% of patients, with a high incidence of serous otitis. Sensorineural hearing loss (SNHL) occurs in 10% of patients. The cochlea is sometimes shorter than usual. Ossicular chain anomalies, especially the stapes, may be seen. The lips are broad and dry, and rhagade furrows around the mouth are seen (Fig. 107.8). The tonsils are often hypertrophied and not recessed in the fossae. The palate is narrow. Cleft lip or palate is occasionally seen. Gingivitis, periodontal disease, and dental caries exist. Microdontia, hypoplasia of the enamel, posterior cross-bite, and widely spaced teeth are frequent occurrences. Articulation defects and dysrhythmic or explosive speech are associated with a low-pitched hoarse voice.
Of all cases, 95% are the result of trisomy 21 with 47 chromosomes. Mosaicism is observed in 2.4% of patients.
Translocations account for 3% to 5% of cases involving chromosome 14, 21, or 22 (i.e., 14/21, 21/21, or 22/21). The incidence of the syndrome increases with advanced maternal age, being 1:1,500 for mothers aged 15 to 29 and 1:50 for those older than age 40. The general incidence is 1:600 of all live births (1, 2, 9).
Translocations account for 3% to 5% of cases involving chromosome 14, 21, or 22 (i.e., 14/21, 21/21, or 22/21). The incidence of the syndrome increases with advanced maternal age, being 1:1,500 for mothers aged 15 to 29 and 1:50 for those older than age 40. The general incidence is 1:600 of all live births (1, 2, 9).
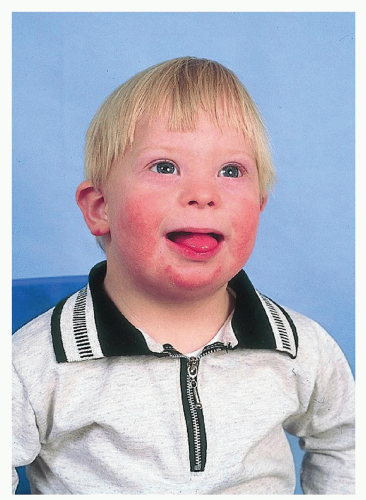 Figure 107.8 Down Syndrome. Reprinted from Pillitteri A. Maternal and child health nursing, 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2003, with permission. |
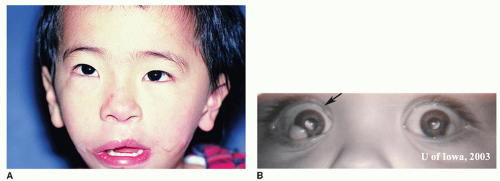 Figure 107.9 A: Patient with Goldenhar syndrome: Note the unilateral microtia, mandibular hypoplasia, and microstomia B subconjunctival dermoids of the right eye in patient with Goldenhar syndrome. A: From Tasman W, Jaeger E. The Wills eye hospital atlas of clinical ophthalmology, 2nd ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2001. B: From Graff JM, Bhola R, Olson RJ. Goldenhar Syndrome (Oculo-Auriculo-Vertebral Spectrum): 6-day-old male with limbal dermoids on http://webeye.ophth.uiowa.edu, with permission. |
GOLDENHAR SYNDROME
Oculoauriculovertebral Dysplasia Spectrum or Facioauriculovertebral Sequence
Gorlin proposed the term oculo-auriculo-vertebral dysplasia for the spectrum of anomalies ranging from hemifacial microsomia (HFM) that denotes unilateral microtia, mandibular hypoplasia, and microstomia to Goldenhar syndrome, which also includes epibulbar dermoids (Fig. 107.9B) and vertebral anomalies. Blepharoptosis, microphthalmia, epibulbar tumors, and retinal abnormalities have been documented. The visual acuity is usually reduced. Mental retardation varies in the reported series between 5% and 15%. Children with anophthalmia or microphthalmia and cleft lip or palate seem at increased risk for cerebral malformation and mental retardation. Various cardiac anomalies are described ranging from ventricular septal defect, patent ductus arteriosus, and Fallot tetralogy to coarctation of the aorta. Renal ectopia and hydronephrosis are less common. Talipes equinovarus deformities and other limb anomalies are rare occurrences. The mandibular ramus and condyles are hypoplastic. The maxilla, malar, and temporal bone are smaller on one side (Fig. 107.9A). The mastoid is also poorly pneumatized. Approximately one-third of the cases show bilateral involvement. The right side is involved in 60% of the cases. Macrostomia or pseudomacrostomia (lateral cleftlike extension of the corner of the mouth) is a usual occurrence. The parotid gland is also affected (agenesis or displaced salivary tissue). Cleft palate or lip occurs approximately 10% of the time. Delayed dental development, as well as lingual and palatal malformations, occurs
on the hypoplastic side. Microtia or occasional anotia and preauricular tags occur between the tragus and the corner of the mouth. The middle ear demonstrates ossicular anomalies and abnormal course of the facial nerve. The hearing loss is more commonly conductive than sensorineural. Cervical vertebral anomalies occur in 30% of cases. Hemivertebrae and fused vertebrae have been described. Tracheal and pulmonary anomalies and TE fistula are less common (1, 10



on the hypoplastic side. Microtia or occasional anotia and preauricular tags occur between the tragus and the corner of the mouth. The middle ear demonstrates ossicular anomalies and abnormal course of the facial nerve. The hearing loss is more commonly conductive than sensorineural. Cervical vertebral anomalies occur in 30% of cases. Hemivertebrae and fused vertebrae have been described. Tracheal and pulmonary anomalies and TE fistula are less common (1, 10
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
