Fig. 4.1
A buphthalmic eye in a case of neurofibromatosis typeie type 1. The sclera is very distensible in the infant eye, and the eye becomes uniformly enlarged due to increased pressure with thinning of the sclera. In addition there has been corneal trauma with a fibrous ingrowth, cataract formation and retinal detachment (scale bar 1 cm)
In adults, the sclera is poorly distensible, and ectasias (local protrusion of thinned sclera) or staphylomas (scleral thinning with uveal show) may occur after trauma or inflammation (Fig. 4.2).
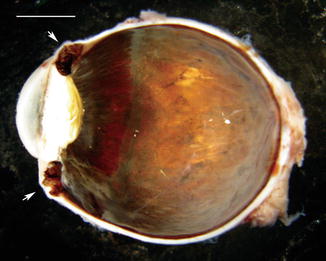
Fig. 4.2
Anterior staphyloma. In a case of long-standing childhood glaucoma, the globe is enlarged, and there is marked thinning of the sclera anteriorly (arrows). The lens also contains a cataract (scale bar 1 cm)
4.3 Congenital Abnormalities
4.3.1 Synophthalmia and Cyclopia
Definition
This is the partial (synophthalmia) or total (cyclopia) union of the elements of the two eyes in the midline region of the forehead to form a single structure.
Epidemiology
The true incidence of cyclopia and synophthalmia in humans is unknown, but it has been estimated at around 1 in 40,000 births.
Aetiology
Clinical Features and Pathology
Abnormal formation of the mesoderm between the optic vesicles leads to partial (synophthalmia) or complete (cyclopia) of the eyes. This usually occurs in association with malformation of the anterior neural plate. In synophthalmia there will be two corneas, two lenses and parts of the iris, ciliary body and retina (Fig. 4.3) [8]. There may be a fused medial sclera, or the midline sclera and uveal tissue may be absent [9] (see also page 17).
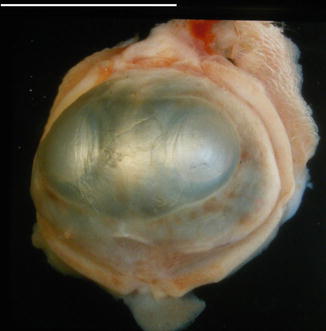
Fig. 4.3
A synophthalmic eye with two corneas and an enlarged scleral envelope (scale bar 1 cm)
4.3.2 Microphthalmos and Nanophthalmos
Definition
Microphthalmos is a condition in which the eye is smaller than normal and may be associated with other developmental defects. Nanophthalmos is used when the eye is microphthalmic but otherwise normal.
Epidemiology
Microphthalmos and nanophthalmos have been reported to occur in up to 30 per 100,000 of the population. Microphthalmia occurs in up to 11 % of blind children.
Aetiology
Microphthalmia is associated with chromosomal abnormalities and single-gene disorders. Many genes have been implicated. It has also been associated with foetal alcohol syndrome and various maternal infections including rubella and cytomegalovirus.
Clinical Features and Pathology
In both microphthalmos and nanophthalmos, the sclera is abnormally thick. As previously mentioned, additional developmental defects including retinal dysplasia may be present in microphthalmos. In nanophthalmos the eye appears normal in structure, but the morphology of the sclera is abnormal. The collagen is unusually thickened, and the bundles are irregularly interlacing [10]. Biochemical studies have shown a loss of chondroitin sulphate proteoglycan compared with controls [11] (see also page 16).
4.3.3 Osteogenesis Imperfecta
Definition
Osteogenesis imperfecta is a congenital bone disorder.
Synonyms
This condition is also known as brittle bone disease or Lobstein syndrome.
Epidemiology
The estimated incidence of osteogenesis imperfecta is 1 in 20,000 births.
Aetiology
The osteogenesis imperfecta group of genetic disorders predominantly affects the bones, and sufferers are prone to bones that break easily following mild trauma. There are at least 8 recognised forms of varying severity [12, 13]. Osteogenesis imperfecta is most commonly caused by mutation in genes encoding the alpha-1 and alpha-2 chains of type 1 collagen or proteins involved in posttranslational modification of type 1 collagen. The most common mutation is autosomally dominantly inherited and due to a null allele in the COL1A1 gene [13].
Clinical Features and Macroscopy
The mildest form (type I) is characterised by a blue-grey tint to the sclera [14]. Scleral abnormalities occur to a lesser extent in other forms of the disease. The sclera is thin and translucent allowing the underlying uvea to become partially visible.
4.3.4 Ochronosis
Definition
This is a syndrome caused by the accumulation of homogentisic acid in the connective tissues which is usually associated with underlying alkaptonuria.
Epidemiology
Alkaptonuria occurs in 1:100,000 to 1:250,000 live births in most European countries. The incidence is very high in Slovakia, 1:19,000.
Aetiology
This is an autosomal recessive condition due to homogentisic acid oxidase deficiency due to mutations on chromosome 3q21-q23. This enzyme deficiency affects tyrosine metabolism resulting in accumulation of homogentisic acid in collagenous structures [17].
Clinical Features and Macroscopy
Progressive pigmentation of the sclera, along with the skin, auricular cartilage and large joints, may occur in alkaptonuria.
4.3.5 Systemic Mucopolysaccharidosis (MPS)
Definition
The systemic mucopolysaccharidoses are a group of metabolic disorders caused by the absence of malfunction of lysosomal enzymes required to break down glycosaminoglycans.
Synonyms
Hurler syndrome, Hurler-Scheie syndrome, Scheie syndrome, Hunter syndrome, Sanfilippo syndrome, Morquio syndrome, Maroteaux-Lamy syndrome, Sly syndrome, Natowicz syndrome
Epidemiology
The incidence varies for the many different types and subtypes. Some subtypes are lethal; others are compatible with longer-term survival. The incidence varies from around 1:250,000 for Hunter syndrome to 1 in 75,000 for Morquio syndrome.
Aetiology
The mucopolysaccharidoses are autosomally recessive disorders due to mutation in different lysosomal enzymes.
Clinical Features and Pathology
The sclera may be thickened due to deposition of glycosaminoglycans [20, 21]. This has been reported in both Hunter syndrome (MPS type II) [21, 22] and Maroteaux-Lamy syndrome (MPS type VI) [23]. Bilateral uveal effusions have been reported in a patient with Hunter syndrome. This was presumed to be secondary to scleral thickening and reduced number of vortex veins [24]. It is possible that the thickening of sclera also contributes to optic nerve swelling.
4.3.6 Ocular Melanosis and Oculodermal Melanocytosis
Definition
This is a congenital disorder resulting in pigmentation of the ocular structures (ocular melanosis) and surrounding skin (oculodermal melanocytosis).
Synonyms
Naevus of Ota
Epidemiology
Ocular melanosis occurs in around 4 per 1,000 Caucasians. Oculodermal melanocytosis occurs in around 3 per 1,000 Caucasians. The incidence is much higher in the Chinese population.
Aetiology
The aetiology is unknown.
Clinical Features and Pathology
In congenital oculodermal melanocytosis, there is slate-grey pigmentation of the sclera (Fig. 4.4). This is due to collections of heavily pigmented melanocytes in the uveal tract, but pigmented dendritic melanocytes are also identified within the sclera [25].
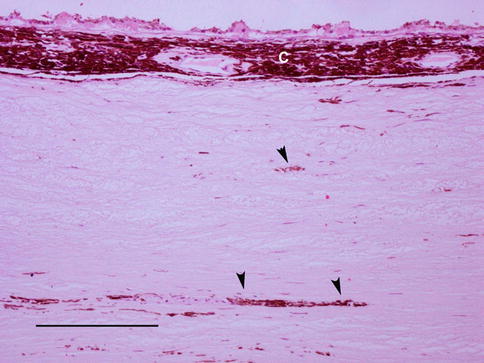
Fig. 4.4
An enucleation specimen from a patient with oculodermal melanocytosis. There is hyperpigmentation of the choroid (c) with patchier pigmentation of the sclera (arrowheads) (scale bar 100 μm)
Prognosis and Predictive Factors
There is an increased risk of uveal melanoma in these patients.
4.4 Inflammation
4.4.1 Episcleritis
Definition
Inflammation occurring in the episclera, a thin layer of tissue covering the sclera.
Epidemiology
It affects all age groups but most commonly occurs in middle-aged women. As initially stated, the majority of cases are isolated, but between 26 and 32 % of cases may be associated with systemic disease or be the herald of a systemic disease [26].
Aetiology
Diseases associated with episcleritis show crossover with those associated with scleritis (see later) and include vasculitides, connective tissue disease, inflammatory bowel disease and relapsing polychondritis.
Clinical Features and Macroscopy
Isolated inflammation of the episclera is usually benign and self-limiting and may be diffuse, sectoral or nodular.
Diffuse and Sectoral
Either diffuse or sectoral redness of the eye may occur sometimes associated with pain and tearing.
Nodular
This presents as small, usually single, episcleral nodules with overlying dilated conjunctival vessels. These are more commonly associated with underlying diseases, particularly rheumatoid arthritis.
Histopathology
Since it is benign and self-limiting, biopsy of episcleritis is uncommon. Diffuse and sectoral episcleritis will show vascular dilatation and congestion with a perivascular lymphocytic infiltrate and variable stromal oedema.
Histology of nodular episcleritis reveals a non-granulomatous, non-specific inflammatory process. The presence of necrobiotic granulomas or a vasculitis should alert the pathologist to the possibility of underlying disease such as rheumatoid arthritis or Wegener’s granulomatosis.
Prognosis and Predictive factors
As previously stated, this is usually a benign and self-limiting condition, and ocular complications resulting from episcleritis are less frequent than that with scleritis.
4.4.2 Scleritis
Definition
Inflammation occurring in the sclera.
Epidemiology
Scleritis is not a particularly common disease. It tends to be more common in women and is most common in the fourth to sixth decades of life.
Aetiology
Inflammatory disease of the sclera may be primary and confined to the eye or seen in association with systemic disease. The incidence of systemic disease is reported to be between 39 and 50 % [27–29]. Scleritis may involve the anterior or posterior sclera, and concurrent and depending on the cause, sequential involvement of both the anterior and posterior sclera may occur [27]. Scleritis is most commonly associated with rheumatoid disease, but there is a broad differential diagnosis [27, 28].
Autoimmune Disease
Rheumatoid Arthritis – 17–33 % of patients with scleritis have rheumatoid arthritis. By contrast only 0.2–6.3 % of patients with rheumatoid arthritis have scleritis [30]. Scleritis may be associated with peripheral ulcerative keratitis. Rheumatoid factor is positive in 60–80 % of these patients.
Wegener’s Granulomatosis – Ocular involvement occurs in around 50 % of patients with Wegener’s granulomatosis [31]. Orbital involvement is more common, but scleritis occurs in around 7–10 % of patients particularly in association with peripheral ulcerative keratitis [31]. Serum c-ANCA is raised in the majority of patients.
Relapsing Polychondritis – Ocular symptoms occur in up to 65 % of patients with relapsing polychondritis at some point during the course of the disease [32]. Episcleritis and scleritis are the most common ocular manifestations occurring in approximately 47 % of patients.
Systemic Lupus Erythematosus – Ocular manifestations are relatively common in systemic lupus erythematosus. Scleritis may occur and be isolated or in association with episcleritis [33]. Antinuclear antibodies may be positive, and anti-double-stranded DNA is present in 30–50 % of patients.
Polyarteritis Nodosa – Ophthalmic manifestations are less common in polyarteritis nodosa than in Wegener’s granulomatosis occurring in around 10 % of patients [34]. Episcleritis is more common but severe progressive scleritis may occur, and scleritis may occur in association with ulcerative keratitis [35].
Behçet’s’s Disease – Scleritis has rarely been described in association with Behçet’s disease [36, 37].
Infectious Scleritis
Infectious scleritis is uncommon particularly in the absence of associated keratitis. Infectious scleritis may be caused by virus, bacteria, fungi or parasites.
Herpes zoster is the most common cause of infectious scleritis as part of herpes zoster ophthalmicus [38]. Pseudomonas aeruginosa is the most common postsurgical bacterial infection along with staphylococcal species [39]. Fungal scleritis is common in certain parts of the world usually due to Aspergillus [40, 41]. Acanthamoeba scleritis is usually a secondary consequence of keratitis [42, 43]. Toxoplasma scleritis occurs secondary to the retinochoroiditis [44, 45].
Surgically Induced Scleral Necrosis
Surgically induced necrotising scleritis (SINS) is a local autoimmune reaction known to develop near previous surgical wounds for cataract pterygium, glaucoma, strabismus and retinal detachment [46]. It appears to occur more commonly following postsurgical infection [47]. Usually the scleritis is localised, but surgically induced diffuse scleritis (SIDS) is also recognised [48].
Risedronate-Associated Scleritis
Bisphosphonate drugs are used for the prevention of osteoporosis but may cause ocular inflammation including conjunctivitis and uveitis. Scleritis in association with risedronate therapy has also been described [49].
Irradiation-Induced Scleritis
This is rare because the sclera is radioresistant and scleral necrosis is a rare complication of radiotherapy for uveal melanoma. It has been described in association with plaque brachytherapy [50] and very occasionally following proton beam therapy usually in association with an underlying inflammatory or systemic disease [51].
Clinical Features, Macroscopy and Histopathology
Scleritis is usually classified into anterior scleritis or posterior scleritis (occurring posterior to the equator of the eye). The onset of scleritis is often gradual, the main symptom being severe pain which may be exacerbated by eye movement and is often worse at night. Patients with anterior scleritis may develop a visible area of redness and tenderness. Posterior scleritis may be asymptomatic although pain and visual disturbance are common. The inflammation in the sclera may result in exudative retinal and choroidal detachment, chorioretinal inflammation and disc swelling. This can occasionally be mistaken clinically for an intraocular tumour (Fig. 4.5) [52, 53]. Severe swelling of the sclera and retro-ocular tissues can produce proptosis.
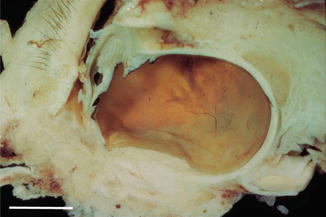
Fig. 4.5
An exenteration specimen showing a nodular posterior scleritis forming a mass lesion with exudative retinal detachment mistaken for melanoma (scale bar 1 cm)
Anterior Scleritis
Anterior scleritis (scleritis occurring anterior to the equator) is more common than posterior scleritis because of the more abundant anterior vascular supply. There are three clinical subtypes of anterior scleritis, diffuse, nodular and necrotising, and most patients’ disease remains in that subtype throughout the course of their disease.
Diffuse Anterior Scleritis
Diffuse anterior scleritis is characterised by severe pain often in the distribution of the trigeminal nerve with redness and swelling of a sector of sclera and episclera. The condition is bilateral in about 50 % of cases. Specimens are rarely submitted to the histology laboratory.
Nodular Anterior Scleritis
Nodular anterior scleritis is characterised by well-defined nodules in the sclera which are not mobile and may be multiple. The overlying episclera and conjunctiva may be inflamed and congested but are separate from these nodules.
Necrotising Anterior Scleritis
Necrotising anterior scleritis may occur with or without inflammation.
With inflammation: When it occurs with inflammation, it is severely painful and destructive and is bilateral in about 50 % of cases. There is an area of severe scleral oedema and congestion, and the overlying episclera may be avascular. The process usually starts in one area and may then spread circumferentially around the globe to involve the whole of the anterior segment.
Without inflammation: Unlike necrotising anterior scleritis with inflammation, this process is rarely painful. There is a grey or yellow patch on the anterior sclera which represents an area of necrosis. This may eventually slough from the underlying sclera, leaving the choroid covered by only a thin layer of conjunctiva as in scleromalacia perforans (Fig. 4.6).
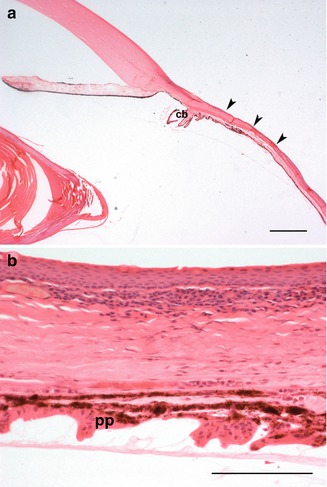
Fig. 4.6
(a) In scleromalacia perforans, there is marked thinning of the anterior sclera (arrowheads). The ciliary body (cb) is stretched (scale bar 100 μm). (b) A higher-power view of scleromalacia perforans. The pars plicata (PP) of the ciliary body is stretched. Inflammation is minimal (scale bar 100 μm)
Posterior Scleritis
Posterior scleritis may occur in association with anterior scleritis or may be isolated. Posterior scleritis is usually unilateral. In addition to pain there may be secondary inflammation in the choroid with exudative retinal detachment and optic disc swelling. Diffuse and nodular forms are also recognised.
Diffuse Posterior Scleritis
In diffuse posterior scleritis, a large area of scleral collagen is thickened by the inflammatory process, the so-called brawny scleritis (Fig. 4.7). There may be large areas of necrotic scleral collagen surrounded by granulomatous inflammation (Fig. 4.8). Alternatively, there may be diffuse granulomatous or non-granulomatous inflammation without areas of necrosis. As this is often a chronic process, there may be associated reactive fibrosis, and the sclera becomes massively thickened. As the process burns out, there may be scleral thinning resulting in staphyloma formation.
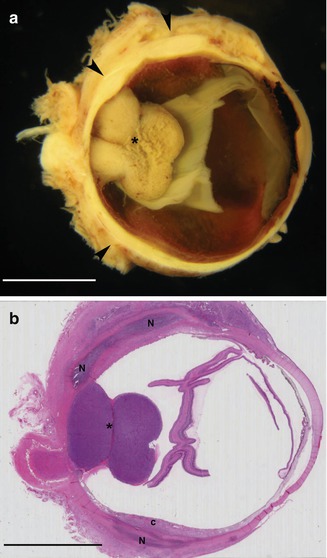
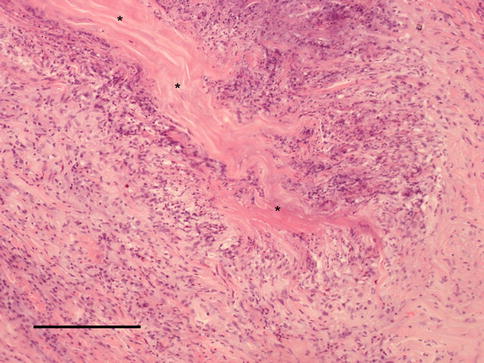
Fig. 4.7
(a) This eye with malignant melanoma developed a diffuse posterior scleritis. The posterior sclera is irregularly thickened (scale bar 1 cm). (b) Histology of this eye shows large areas of scleral necrosis (N) not related to the melanoma. The choroid (c) is secondarily involved by inflammation (scale bar 1,000 μm)
Fig. 4.8
Posterior scleritis showing a zone of necrobiotic collagen surrounded by a palisaded granulomatous reaction (scale bar 100 μm)
Nodular Posterior Scleritis
In nodular posterior scleritis, the inflammatory process forms a localised mass around an area of collagen necrosis. In nodular scleritis there is usually a discrete zone of necrotic scleral collagen surrounded by granulomatous inflammation.
In any form of scleritis, the presence of vasculitis in association with scleritis is in keeping with a systemic immune-mediated condition such as Wegener’s granulomatosis or rheumatoid disease (Fig. 4.9). The presence of microabscesses in areas of scleral collagen necrosis should prompt a search for infectious organisms (Fig. 4.10).
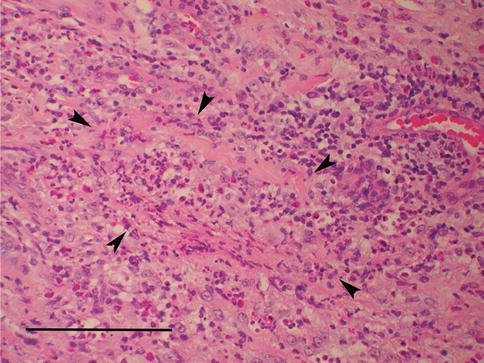
Fig. 4.9




Small blood vessels (arrowheads) with inflammatory infiltration of the wall and occlusion of the lumen in Wegener’s granulomatosis (scale bar 100 μm)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
