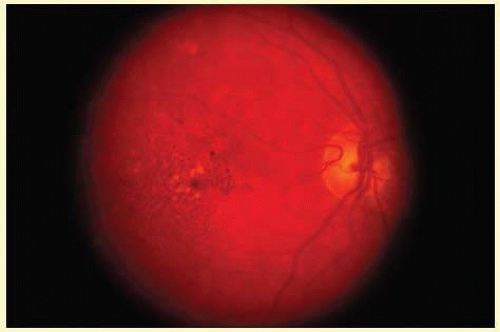
 FIG. 23C.1 Drusen. Fundus photograph demonstrates numerous small drusen and RPE pigment clumping in an eye with “dry” AMD. |
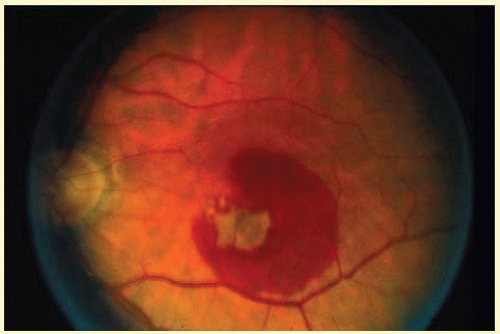 FIG. 23C.2 Exudative AMD. A large neovascular membrane with associated subretinal hemorrhage consistent with “wet” or exudative AMD is present in the macular region. |
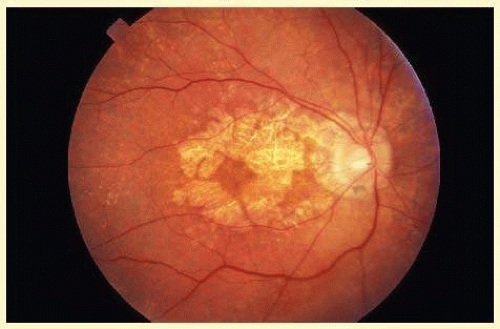 FIG. 23C.3 Atrophic AMD. Choroidal and RPE atrophy surround the optic disc and spare the foveal region in this example of atrophic AMD. |
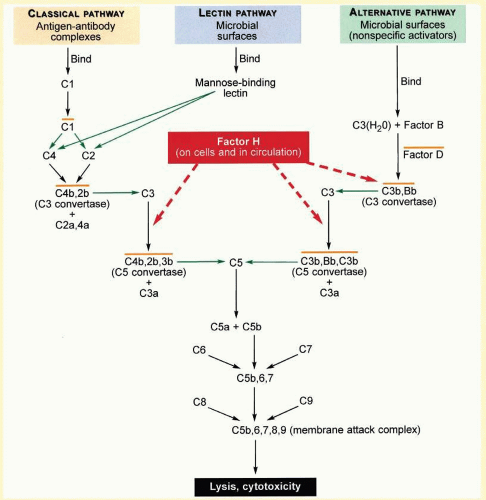 FIG. 23C.4 An Overview of Inflammation and the Complement System. Green arrows indicate proteolytic cleavage of the molecule at the tip of the arrow. Orange lines over complexes indicate that each is enzymatically active. Dashed red arrows indicate the roles of factor H in binding to C3b, accelerating the decay of the alternative pathway C3-convertase (C3bBb) and acting as a cofactor in proteolytic inactivation of C3b. (Modified from Levinson W, Jawetz E. Medical Microbiology and Immunology. 7th ed. New York, NY: The McGraw-Hill Companies; 2002:401.) The complement system is composed of a group of inactive plasma enzymes (zymogens) and cell membrane regulatory proteins that interact to maintain a balance between activation (with phagocytosis of pathogens) and inactivation (with protection of self). In response to initiating stimuli, the complement cascade produces peptide mediators of inflammation, phagocyte recruitment, opsonization, and cell lysis. The three pathways lead to complement activation. They differ in their initiating stimuli. Once activated, the complement reaction in most cases takes place on the cell surface on which it was initiated. The three pathways converge on complement C3b, the primary effector of the complement system. The classical pathway is activated by antigen-antibody complexes on the surface of invading pathogens. The mannan-binding lectin pathway is activated by pathogen mannose and is important in infancy when adaptive immune system is not yet able to produce antibody. The alternative pathway is initiated spontaneously and continuously in the plasma where complement C3 undergoes spontaneous hydrolysis. As factors D and B cleave and activate the C3 convertase, complement C3b is produced and deposited on both pathogens and host cells. Complement C2 is an activator of the classical pathway analogous to factor B. C3b opsonizes cells marking them for phagocytosis by cells with complement receptor Cr1. Other C3 cleavage products, C3a and C5a, are potent mediators of both a local inflammatory response and phagocyte recruitment and in addition, have been shown to promote choroidal neovascularization by increasing VEGF expression in mice.205 The terminal peptides, C5b, C6, C7, C8, and C9 interact to form the membrane attack complex (MAC) and induce cell lysis. Excessive amplification of the complement system is regulated by additional proteins. In the alternative pathway, MCP (membrane cofactor protein) and DAF (decay accelerating factor) are the cell-bound proteins that prevent complement activation on host cells, and plasma proteins factor H and factor I down-regulate inflammation by coating host cells marked with C3b and inactivating plasma C3b respectively. The CX3CR1 receptor for the chemokine, fractalkine, a membrane bound protein is expressed by endothelial cells, neurons, and glia. Its soluble form, released by TNF-alpha-converting enzyme (TACE), is important in the adhesion and recruitment of monocytes, natural killer cells (NK), and activated T cells which bear the CX3CR1 receptor. Fractalkine blocks TNF-alpha secretion and thereby participates in the downregulation of inflammation. |
TABLE 23C-1 Components of Drusen | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 23C-2 Nonocular Diseases with Drusen-like Deposits | ||||||||
|---|---|---|---|---|---|---|---|---|
|
TABLE 23C-3 SNPs Associated with AMD | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


