The Major Histocompatability Complex
Rahul Mandiga
Tracy Gosen
Varsha Manjunath
Stefan D. Trocmé
OVERVIEW
The major histocompatability complex (MHC) was initially defined on the basis of mapping gene loci that were the most significant determinants of acute graft rejection, in contrast to the minor histocompatability genes involved in chronic rejection.1 Important groundwork for the discovery of MHC was laid in the early 1900s with allogenic tumor transplantation studies and Landsteiner’s discovery of an agglutination phenomenon that led to ABO blood typing.2,3,4 Understanding of MHC evolved in the 1950s, when leukocyte agglutinating antibodies were first found in the sera of multiply transfused patients and in the sera of up to 30% of multiparous women. Serologic reaction patterns showed that each antiserum reacted with leukocytes from many but not all individuals. These patterns indicated that antibodies in the sera were reacting to a polymorphic system of alloantigens, antigens that vary among members of the same species. The concept of a major histocompatability gene was first introduced in 1956 by Snell et al.4
As human MHC molecules were first found on white blood cells, they were named human leukocyte antigens (HLA). Later these antigens were identified on virtually all nucleated cells throughout the body, including cells in all three layers of the cornea.5 In the 1960s, differentiation of different HLA was made possible by the use of serologic reagents to identify antigenic determinants, also called epitopes, the binding site for antibodies on an antigen.6 International collaboration commenced, and with it the need for a uniform method for identifying and naming HLA. MHC restriction, T-cell recognition of a viral antigen being restricted by MHC, was first recognized in 1974. However, it was another 10 years until the relationship between MHC restriction and T-cell–mediated immune responses was proven. By 1977, it was established that class I would describe HLA-A, HLA-B, and HLA-C antigens and class II would be used for HLA-DR, HLA-DQ, and HLA-DP.4,7 Soon thereafter, it was discovered through multicenter trials that patients with the best possible match of HLA-A, HLA-B, and HLA-DR had more favorable outcomes after kidney transplantation.
In 1973, HLA-B27 was found to be reproducibly associated with ankylosing spondylitis, with a relative risk of greater than 100, providing an impetus for further study of HLA in autoimmune diseases. By defining HLA disease associations, at-risk populations and genetic susceptibility can be identified. Associations have shed light on the mechanisms of disease, which include autoimmune, immune complex, and nonimmune, allowing for more precise strategies in disease prevention and treatment.4 This topic is discussed in detail in a following section.
HLA classification has been refined by technologic advances, such as microcytotoxicity testing, electrophoresis, and DNA sequencing, while computer codification of anti-HLA alloantisera reaction patterns have made possible the delineation of the HLA system.8 International collaboration has continued to be a major part of this field, with a central agency for reporting new alleles and monthly publications of the updated HLA sequence database held at the Anthony Nolan Research Institute.9
In this chapter, nomenclature of the HLA system, genetic organization, biochemistry, and structure are reviewed. The historic and modern techniques of HLA typing are discussed, as well as the ophthalmic diseases associated with the HLA system.
NOMENCLATURE
The MHC HLA region is located on the short arm of chromosome 6, 6p21.3, and contains greater than four megabases, making up more than 220 polymorphic genes of diverse function (Fig. 38-1).10 MHC loci are categorized into three classes, according to function. With more sensitive serologic determination, DNA sequencing, and other technologies applied to the field, many allele sequences have been refined and renamed. The HLA Nomenclature Committee, governed by the World Health Organization, has met every 2 to 3 years since 1964 to address the evolving knowledge and to standardize the criteria used for defining serologic determinants.11 Additionally, new alleles are amended to the HLA/IMGT (immunogenetics) database and verified to ensure that added alleles are in fact different. Alleles named in error or found to be duplicates are renamed or abandoned.12 As of October 2009, a total of 4,270 HLA and related alleles have been determined, and new alleles are updated monthly at a rate of about 450 sequences per year. The complete, updated database can be accessed on the Internet (http://hla.alleles.org).10 Additional loci continue to be recognized, and several are apparently silent (i.e., no gene product is produced). Despite its complexity, the HLA nomenclature system has been a prototype for naming genes in the Human Genome Project.11
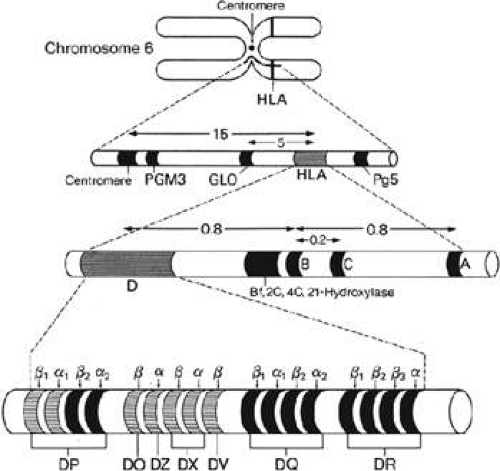 Figure 38-1. The genetic organization of the MHC on chromosome 6. Note the relation of the HLA loci relative to each other and in relation to the centromere. A complete database of nucleotide sequences within the MHC is available from the Trowsdale Library. (Figure by Varsha Manjunath adapted from previous edition.) |
ALLELIC POLYMORPHISM
At any given locus within the MHC complex, one of several allelic variants of a gene can be found. The high degree of polymorphism exhibited by MHC genes has been attributed to the rate of mutation of HLA. Immunologically, the high degree of polymorphism is important for covering a vast array of antigens that need to be presented to T cells.13 There are three methods for the generation of new classical MHC gene sequences: point mutation, recombination, and microrecombination. Point mutations are single nucleotide alterations, recombination occurs when a molecule of nucleic acid is broken and joined to a different strand, and microrecombination is a small scale of recombination, genetic information exchange within a gene.
One example of this is HLA-A69, whose sequence is a composite of the 5′ sequence of HLA-A68 containing the α1 domain and the 3′sequence of HLA-A2 containing the α2 and α3 domain, clearly resulting from a reciprocal recombination event. Given the homology of the α2 subunits, the PA2.1 antibody binds with the same affinity to HLA-A69 and HLA-A2.14 This serologic similarity highlights the necessity of vigorous testing, including DNA sequencing, in the identification of a new HLA.9
PUBLIC AND PRIVATE EPITOPES
The public and private antigens terminology was introduced in the early days of serologic testing. Early antisera were often nonspecific and recognized an epitope on several different molecules, known as public epitopes. Development of more specific antisera allowed for greater discrimination between epitopes, known as private epitopes. For example, the early identified DR5, a public epitope, was actually a composite of DR11 and DR12, private epitopes.6
CROSS-REACTIVE EPITOPE GROUPS (CREGs)
HLA can be grouped on the basis of a public epitope, which arises from sharing of polymorphisms among different HLA types. CREGs have been investigated as an alternative to HLA matching; however, there is controversy over this issue.15,16,17 It has been proposed that the CREG classification definition and DR distribution impacts the graft survival.18 CREGs have also been investigated as a means of matching to decrease discrepancies of allocation to Caucasians over minorities.15,17
GENETIC ORGANIZATION
Humans inherit one chromosome 6 from each parent; each chromosome has an HLA haploid genotype, or haplotype.19 Because all HLA genes are codominant, both alleles at a given HLA locus are expressed and their products are detected on the cell surface. This codominance creates a greater variety of MHC molecules, which allows for a better immune response to a diverse array of pathogens. By classic Mendelian inheritance, there is a 25% chance that two siblings will share both haplotypes, a 50% chance that they will share one haplotype, and a 25% chance that they will not share either haplotype.20,21 These calculations are strictly based on Mendelian genetics and do not take into account recombination, particularly hot spots in the class II region, resulting in allelic and haplotypic diversity.19 Considering the large number of gene loci in the MHC, and a significant amount of polymorphism at each locus, a normal population will have many different haplotypes or combinations of HLA-A, -B, -C, -DR, -DQ, and -DP.
LINKAGE DISEQUILIBRIUM
Linkage disequilibrium is the nonrandom association of alleles at two or more loci, either with greater or lesser frequency than would be expected with the random formation of haplotypes. Certain combinations of HLA alleles are found with a frequency far exceeding the expected rate.19 For example, the HLA-A1 allele and the HLA-B8 allele have been demonstrated in American Caucasians at frequencies of 27.5% and 15.7%, respectively. Given this, the expected frequency of the HLA-A1/B8 haplotype is 0.275 × 0.157 = 4.3%. However, this haplotype is actually found with a frequency of about 9.8%, making the linkage disequilibrium 9.8 – 4.3 = 5.5%. This indicates a low frequency of recombination between HLA-A and HLA-B.22 Etiologies of linkage disequilibrium in human populations include inbreeding, migration, and mixing of two populations, random drift, and selective advantage of certain haplotypes.23
Clinically, linkage disequilibrium can lead to an association of both HLA alleles with a disease process. Autoimmune diseases are frequently associated with HLA loci, typically class II, and vary between ethnic groups. For example, Sjögren’s syndrome (SS) has been associated with haplotypes B8, DRw52, and DR3 in populations of European descent; DR2 in Scandinavians; and DR5 in Greeks.24,25 A similar situation has been demonstrated in Graves’ disease and diabetes mellitus.26,27
HLA COMPLEX AND PRODUCT STRUCTURE
The human MHC complex is divided into three regions. Farthest from the chromosome’s centromere is the class I region, which is the largest and contains the most gene fragments. Besides its role in adaptive immunity, class I molecules are involved in IgG uptake in the gut, regulation of iron metabolism, and regulation of natural killer (NK) cell function.13,28 The class II region is at the other end of the complex and is more compact and almost entirely dedicated to genes involved in antigen processing and presentation. The class III region, or central MHC, does not contain any class I or class II genes. The invariant light chain molecule and the class I HLA β2-microglobulin are located on chromosome 15.13 Both class I and class II MHC molecules are heterodimeric (two polypeptide chains) glycoprotein cell-surface molecules (Fig. 38-2) whose structures were determined by x-ray crystallography.29
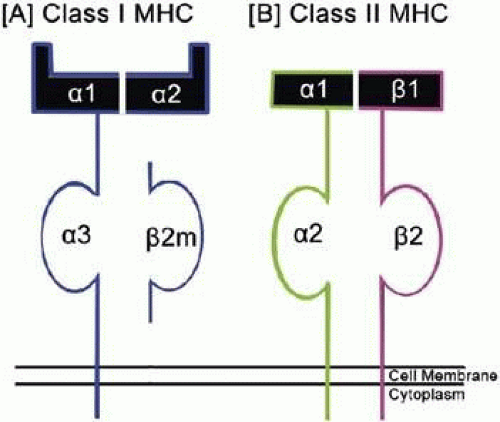 Figure 38-2. A schematic presentation of the structure of (A) class I MHC and (B) class II MHC molecules. (Figure by Varsha Manjunath.) |
CLASS I REGION AND STRUCTURE
The class I region encodes for three main glycoproteins: A, B, and C. It consists of seven exons (translated regions of DNA), separated by introns (untranslated regions of DNA). The second, third, and fourth exons correspond to the α1, α2, and α3 heavy chain domains, respectively.14 Inherently, complex mechanisms for gene splicing, assembly, and ribonucleic acid (RNA) translation are required to produce intact HLA molecules.
Evaluation of the chromatin organization of chromosome 6 has helped elucidate the structure and function of class I molecules.30 Class I HLA consist of a glycoprotein heavy chain with three domains (α1, α2, and α3) on the cell surface and a non–covalently associated protein, β2-microglobulin.28 The entire class I molecule is anchored into the cell membrane lipid bilayer via the α3 domain and the β2-microglobulin.31 The α1 and α2 heavy chain subunits determine the specificity of the particular HLA, forming the peptide binding site. The peptide binding groove limits the length of the presented peptide since the binding site must interact with the N- and C-termini of the peptide. The peptides can vary from eight to ten amino acids in length, but are predominantly nonamers. The most important MHC interactions are those with side chains of the second amino acid of the peptide (referred to as P2) and the final amino acid, indicating that these sites have the greatest influence on the specificity of the interaction.1,21
CLASS II REGION AND STRUCTURE
The map of the class II region (Fig. 38-1) shows a series of subregions, each containing A and B genes encoding α and β, respectively.21 The DP, DQ, and DR genes, like class I genes, are composed of introns and exons, with the exons corresponding roughly to the protein domains.22 The DR subregion, for example, contains a single DR-A gene and up to nine DR-B genes.32 Similarly, the DP and DQ subregions each contain one expressed α gene and β gene, and additional unexpressed pseudogenes.21
The HLA class II region encodes three highly polymorphic glycoproteins, DP, DQ, and DR, and two oligomorphic glycoproteins, DO and DM. Each of the class II glycoproteins is a non-covalently linked heterodimer with two transmembrane chains (α and β) (Fig. 38-2). These chains each consist of three regions including a cytoplasmic region, a short transmembrane region, and an extracellular region. The cytoplasmic portion can be phosphorylated for signal transduction. The extracellular region is composed of two domains, α1 and α2, or β1 and β2.21 The α2 and β2 domains near the cell surface show significant sequence homology to immunoglobulin constant region domains. The α1 and β1 domains contain the binding groove. In class II molecules, the binding site is not a pocket at each end, so the peptide can extend beyond the edges of the groove. This allows peptides binding to class II molecules to be larger than those bound by class I molecules, generally 13 to 25 amino acids, but can be much longer.13
CLASS III REGION
The class III region contains several clusters of genes, though none that encode for class I or class II molecules. This region contains genes related to the immune system, such as C2, C4, and factor B of the complement pathway, two heat shock proteins, tumor necrosis factor (TNF) α and β, 21-hydroxylase.21 There are also proteins without known roles in the immune system coded here, such as NOTCH4, PPT1, and steroid 21-hydroxylase.34,35
HLA DISTRIBUTION AND FUNCTION
Class I Molecules
As previously mentioned, HLA class I molecules (HLA-A, -B, and -C) are expressed on virtually all nucleated cells, including all three layers of the immunoprivileged cornea.5,36
Class I molecules display endogenous antigens processed through the cytosolic pathway. In virus-infected cells, the class I molecules present viral peptides to activate CD8+ cytotoxic T lymphocytes (CTLs).37 The infected cell breaks down the viral proteins into peptides using proteasomes in the cytoplasm, which are then carried into the rough endoplasmic reticulum (RER) by transporter proteins (TAP1 and TAP2) (Fig. 38-3). The class I MHC α-chain is folded and associated with β2-microglobulin, and the appropriate peptide is loaded into the binding groove. The complex is then transported from the RER to the Golgi and finally to the cell surface. T-cell receptors (TCRs) will recognize a peptide epitope only in the context of an MHC molecule complex, which means that free viral peptides in the extracellular space cannot activate TCRs.38,39
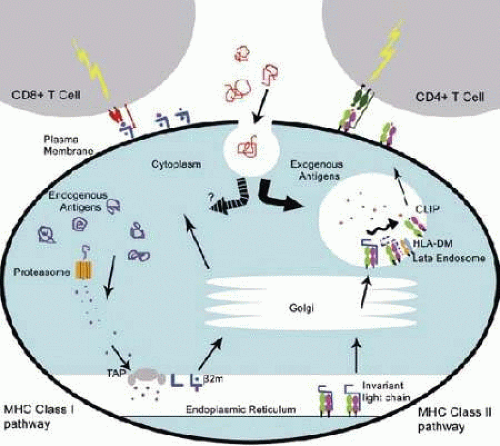 Figure 38-3. MHC class I and MHC class II presentation pathways. On the left, the MHC class I pathway is depicted, where cytosolic or endogenous proteins are broken down into peptides by proteasomes and transported by TAP into the rough endoplasmic reticulum. The proteins are loaded onto the MHC and transported to the Golgi and then to the plasma membrane for presentation. On the right, the MHC class II pathway is shown, where peptides derived from the extracellular or exogenous environment are held in a vesicle until fusion with the endosome-containing MHC class II molecules can occur. Again, the MHC molecule forms in the rough endoplasmic reticulum, but the binding site is protected by the invariant chain. The MHC class II are always passed down the pathway with HLA-DM, which removes the CLIP, the final remnant of the invariant chain, and allows the exogenous peptide to bind. The final complex is transported to the cell membrane. (Figure by Varsha Manjunath.) |
Interestingly, a TCR is specific for its peptide-MHC complex. The same viral peptide presented by a different class I molecule does not produce a strong enough reaction to activate the CTLs.40 The CD8 coreceptor binds to the α3 domain on the MHC class I heavy chain, acting as an adhesion molecule to allow for a stronger interaction between the peptide–MHC complex and the TCR to enhance subsequent T-cell activation. Recall that the α3 domain has little polymorphism, so the structure varies minimally between class I molecules. If the TCR is activated, the CTL proliferates with the help of IL-2 from the CD4+ T-helper cells and kills the infected cell.41
MHC class I molecules are recognized by the host during tissue transplant rejection. This is demonstrated in cell-mediated cytolysis, which is the experimental correlate of graft rejection.29 Both private and public HLAs are recognized independently by T cells.42,43 For acute rejection, either a donor antigen presenting cell activates a self T cell or a self antigen presenting cell activates a donor T cell to initiate the pathway to eventual cytolysis and graft rejection.13
Class II Molecules
The class II antigens, unlike the class I antigens, are not ubiquitously expressed, but rather, expressed by a small subset of cells called antigen presenting cells (APCs): dendritic cells, macrophages, and B lymphocyte cells. To a lesser extent they are expressed in thymic epithelium and resting T cells.44 Class II molecules DP, DQ, and DR present peptides to CD4+ T cells, the helper T cells (Th cells), which are able to activate the immune system. Class II molecules present exogenous antigens from the extracellular environment, rather than endogenous antigens, which allows for Th cells to be activated by infectious agents in the cellular environment opposed to foreign antigen present within an infected cell. Therefore, antigen internalization and processing is important for obtaining the peptides to be presented by class II molecules.45
The process starts with the synthesis of the class II molecule in the rough endoplasmic reticulum, with a CLIP (class II associated invariant chain peptide) protecting the binding groove (Fig. 38-3). Without the CLIP, the class II molecules would present the same peptides as the class I molecules. The class II molecules are packaged into endosomes with HLA-DM molecules, which assist in removal of the CLIP.46 The endosome fuses with a vesicle created via macropinocytosis, a form of endocytosis by which small particles are brought into the cell. The proteins in the vesicle are broken down to peptides of 12 to 18 amino acids in length, the size needed to fit in the class II binding groove. The class II molecules then bind their associated peptides and move to the cell surface, where they remain for 1 to 2 days.45 The CD4 coreceptor binds to the β2 domain, which holds the peptide-HLA complex in closer proximity of the TCR to allow for tighter binding and subsequent activation and proliferation. Just as in the class I molecules, TCR from CD4+ T cells can recognize only peptides bound to class II molecules.42
HLA ANTIGEN TYPING
It has now been more than 50 years since the vital role of MHC receptors in graft rejection was elucidated, and many laboratory techniques have since been developed in HLA antigen typing to ensure donor–recipient compatibility. Classical serology has been used in the past to phenotype donors and recipients; however, laboratories are increasingly turning to DNA-based methods to directly genotype patients and donors for HLA alleles.47 These new techniques give a better understanding of a patient’s genetic profile and immunogenic risk.48 The following sections outline the low-resolution or serologic methods used historically and the newer high-resolution DNA-based methods.
Serologic Methods
Complement-Dependent Cytotoxicity (CDC) Assay
Developed in the 1960s by Terasaki and McClelland, the CDC assay evaluates alloantibody reactions to HLA on donor cells. Lymphocytes obtained from an individual or cell panels are combined with a potential recipient’s serum (typing sera) and incubated with rabbit complement.49 Anti-HLA antibodies present in the recipient’s serum bind to the donor cell or cell panel, with any bound IgG and IgM antibodies activating the complement cascade to promote cell injury and death.50 Increased membrane permeability of injured cells allows for the uptake of a vital dye, such as trypan blue, eosin, or ethidium bromide, and a quantitative measure of cell viability and cytotoxic effects can then be assessed under phase contrast microscopy. Thus, the CDC assay functions as a screening tool used to detect the presence or absence of anti-HLA antibodies in a rapid and specific manner but does not typically provide any information on HLA specificity of the detected antibodies.51 Only if the HLA phenotype of donor cells is known, can specific alloantibodies be identified.49
Solid-Phase Systems
Recognition that patients with CDC-negative cross-matches still had early antibody-mediated graft rejection52 led to the development of flow cytometry, enzyme-linked immunosorbent assay (ELISA), and multiplex platforms that are more sensitive and specific methods of alloantibody identification and have become gold-standard techniques.21,53,54 The CDC assay lacked sensitivity and specificity since large cell panels were unable to cover the polymorphic HLA system, let alone rare or unusual antigens. Additionally, the assay was unable to discriminate between the existence of HLA class II antibodies in the presence of HLA class I molecules on leukocytes.50 The newest solid-phase techniques overcome the difficulties of the CDC assay through the use of recombinant HLAs produced by EBV-transformed cell lines. Various HLAs and purified HLA class I and class II molecules can be produced, which eliminates false positives due to nonspecific binding.
As a preliminary screening tool, microspheres coated with purified HLAs are incubated with patient sera and analyzed for shifts in fluorescent intensity.51 Once the presence of HLA class I or class II antibodies has been determined, the specificity of the HLA antibody can be determined using specificity assays.
The specificity assay is based on flow cytometry and uses individual microparticles coated with full HLA class I or II type antigens; therefore the binding pattern allows for a panel-reactive antibody percentage and HLA specificity to be determined by software analysis.51 The utility of the method lies in the ability to assay high volumes of samples in a rapid manner. The drawback of this approach arises from the panel used to detect the HLA antibodies. Certain antibody specificities may not be detected due to the masking effect of linkage disequilibrium.
Lymphocyte Cross-Matching
A pretransplantation screening method, cross-matching is used to identify the presence of preformed anti-HLA antibodies in the potential recipient, which can lead to hyperacute graft rejection.21,53,54 Sensitization of the immune system to a great variety of HLAs can occur from previous transplantation, blood transfusion, and pregnancy. The cross-match test is performed using peripheral lymphocytes from the donor, which are separated into B and T cell lines. The serum from the recipient is then mixed with the donor B or T cells. A positive reaction, or induced cell death, indicates the presence of preformed antibodies within the recipient that are reactive to the donor cells. This makes the likelihood of transplant success very low and is therefore an absolute contraindication to the transplantation of major organs.55 To minimize false-positive results, which inherently eliminates potential donors, the recipient’s cells can be cross-matched with their own serum to detect the presence of any nonspecific autolymphocytotoxic antibodies.
Antibody-Mediated Microcytotoxicity Assay
In contrast to the three methods discussed previously, the microcytotoxicity assay is a serologic test used to determine a specific HLA haplotype on donor recipient cells rather than detecting the presence of preformed antibodies. White blood cells from donor and recipient are combined with an array of different antibodies, each directed to specific class I and II MHC molecules. Similar to the CDC, complement is then added and cell damage is evaluated with the use of vital dyes and examined under microscopy. This test detects any cell bearing an HLA allele homologous to the known antibody, and the HLA phenotype can be determined on the basis of the reaction pattern. HLA class I antibodies were identifiable by the microcytotoxicity assay, but HLA class II antibodies often remained unidentified due to the overlapping distribution of MHC class I and II antigens. The test was often not sensitive enough to detect certain alleles that have been detected using DNA-based methods.
Mixed Lymphocyte Reaction (MLR)
The MLR is a unidirectional reaction useful in determining the degree of MHC class II compatibility.56 In this test, a panel of HLA-D homozygous typing cells (HTCs) displaying known HLA-D surface molecules are killed by irradiation or mitomycin-C and serve as stimulator cells. Live responder lymphocytes obtained from the recipient are incubated with the stimulator cells, and the degree of responder cell growth is indicated as a measure of DNA incorporation of [3H] thymidine.54 The greater the MHC II difference between the stimulator and responder cells, the more robust response by the responder lymphocytes in the MLR assay. As a strong positive reaction suggests poor graft outcome, the best match involves responder cells with the least cell growth.
To determine if the responder cell is heterozygous or homozygous for a specific HLA-D type, the reaction can be reversed. The responder lymphocytes from the recipient are killed and used as stimulator cells. When incubated with the live HTC cells, if no DNA synthesis occurs, the recipient’s cell is HLA-D homozygous. However, if DNA synthesis does occur, the HTC is reacting to a foreign HLA-D type not available in the HTC panel, and the recipient’s cell is HLA-D heterozygous.57
DNA-Based Methods
Molecular HLA Typing
The large degree of HLA polymorphism and the need for high-resolution typing has driven the development of sensitive HLA typing to improve transplant outcomes.58 Molecular testing has largely replaced traditional methods due to greater accuracy and cost-effectiveness; however, some laboratories still use both traditional and molecular methods to reach a meaningful result. One of the major drawbacks of serology is the need for viable cells. Blood cells drawn from deceased donors, the usual method for the typing of corneal donors, may have impaired viability and reduced expression of cell surface antigens. This makes serologic typing difficult and prone to errors.59 The use of polymerase chain reaction (PCR) has not only revolutionized the speed of HLA matching, reducing the time needed from several weeks to a few hours, but has also greatly increased the number of known HLA alleles.58 The challenge now is maintaining high-resolution typing with ever-increasing numbers of defined HLA alleles.
Molecular typing of HLA can be performed using DNA or RNA. Restriction fragment length polymorphic DNA probes (RFLPs) are a useful method of typing HLA class II DNA.60 The pattern of DNA fragments for a given HLA gene depends on the gene sequence, the restriction endonuclease used, and the probe used. However, if higher-resolution results are required or an insufficient amount of DNA material is available for RFLPs, PCR techniques have proven to be very useful.
The following are PCR techniques commonly used for HLA typing.58,61,62 Each technique has specific advantages and disadvantages based on resolution, speed, and cost, which dictates which method should be used.63
Probing with sequence-specific oligonucleotide probes (SSOP)
Commercially available SSOP probing is used for the testing of large sample sizes (e.g., bone marrow registry typing). In this technique, sequence-specific probes detect various alleles at a locus using probes recognizing allelic differences and are able to detect the complementary region of an amplified target. The probes are visualized by color reaction or fluorescence. Despite its utility, PCR-SSOP can be slow and cumbersome, making it a poor choice if rapid results are desired.
Sequence-specific primer (SSP)
If a rapid result is required, as in the case of finding an HLA-matched cadaveric donor, then PCR-SSP techniques should be used. In these systems, the amplification process is performed using one primer that is conserved across many alleles and a second primer specific for a one–base pair (bp) difference. This method is based on allele discrimination during PCR amplification. It requires many separate PCRs and more DNA than PCR-SSOP, which makes it a poor choice for rapid throughput of large samples.
RFLPs
This technique uses several different restriction endonucleases with high affinity to specific restriction sites. PCR products containing single nucleotide polymorphisms (SNPs) are digested and run through a gel assay to determine fragment lengths and ascertain whether or not the enzymes cut the expected restriction sites.64 Allele discrimination is achieved using one or two allele-specific probes hybridizing to the SNP polymorphic site that are linked to a fluorophore. This type of testing can be used to resolve ambiguous typing results for other assays, but it is not usually employed for high-throughput needs.
Single-strand conformation polymorphism
The basis of double-stranded sequence conformation polymorphism testing is such that the formation of “loops” and “hairpins” are sequence dependent.65 If two alleles have different sequences, the types of secondary structures should differ. The concept is that DNA sequences allowed to form secondary structures can be discriminated among themselves by the migration of these secondary structures in a gel matrix environment.61 Developments in conformation-based techniques have led to automated RSCA sequencers that use a fluorescein-labeled reference strand, and this methodology could be a potential tool for large-scale typing.63
If HLA matching is crucial at the DNA level, as in the case of hematopoietic progenitor cell transplantation, comprehensive high-resolution HLA sequence-based typing (SBT) genotyping can be used. The chain termination method (Sanger method) and pyrosequencing are two available methods that can be used to determine the order of nucleotides in DNA.66 With the development of automated sequencers and robotic stations, SBT testing can be efficacious and used for high-throughput needs.67
In ophthalmology, postmortem typing can be performed via stimulation of cultured donor retina pigment epithelial cells by interferon gamma. The cells upregulate HLA expression, which can by typed using cytotoxicity and one-dimensional isoelectric focusing.68
HLA AND DISEASE ASSOCIATIONS
The association of particular HLAs to autoimmune and infectious disease susceptibility has been known for a number of years.58,69 Autoimmune diseases are the result of a combination of predisposing genes and precipitating environmental factors. In almost all autoimmune diseases, the strongest genetic predisposition is associated to one or more HLAs. Mapping of the HLA complex genes involved in these diseases and the possible mechanisms behind the associations are therefore now major fields of research.4 The susceptibility to disease, the possible mechanisms of HLA disease association, and the specific diseases associated with HLA and the eye are discussed in the following sections.
GENETIC SUSCEPTIBILITY TO HLA-ASSOCIATED DISEASE
Statistically speaking, the relative risk (RR) describes how much more frequently a disease occurs among individuals carrying an antigen under study compared with those lacking it. It is important to consider that most reported values are based on low-resolution typing and therefore may not be the true RR.63 The absolute risk (AR) is the chance that an individual who possesses the disease-associated HLA will actually develop the disease during his or her lifetime.
Consider the prototypic association between ankylosing spondylitis and HLA-B27. Studies have found that 88% to 96% of the patients with ankylosing spondylitis carry HLA-B27 compared with 8% to 4% of healthy controls.70,71 These data have been confirmed by many groups, giving a RR of greater than 100 to develop ankylosing spondylitis in individuals positive for HLA-B27.
The reason for this very strong association is unknown, but it has been speculated that either immune response genes (IRGs) in strong linkage disequilibrium to HLA-B27 are involved or there was a cross-reaction between HLA-B27 and the etiologic agent causing the disease.4,71
However, despite a very high RR, the AR that an HLA-positive individual will go on to develop an HLA-B27–associated disease is less than 5%. Therefore, it is important to remember that the HLA phenotype is only one part of the overall risk for disease development. Popular hypotheses suggest that an environmental or infectious agent acts as a disease trigger in genetically predisposed individuals.
MECHANISMS OF HLA DISEASE ASSOCIATION
Despite compelling evidence of the association of specific HLA phenotypes with autoimmune diseases, such as ankylosing spondylitis, multiple sclerosis, and birdshot chorioretinopathy, and infectious diseases, such as malaria and HIV, exactly how the HLAs influence disease susceptibility is not yet known. A significant amount of research has been conducted to attempt to elucidate the link, which could aid in treatment of these conditions. The following discussion reviews current theories that support the link between HLAs and disease susceptibility.
Direct Involvement of Human Leukocyte Antigens
Molecular Mimicry
Molecular mimicry is a mechanism by which T cells and antibodies generated against foreign antigens cross-react with self-antigens to initiate an autoimmune response. Infectious agents and molecular mimicry have been postulated to play a role in many autoimmune diseases. An interesting instance of molecular mimicry is the cross-reactivity of anti-DNA antibodies with bacterial antigens and with the EBNA protein of Epstein–Barr virus (EBV).72 High titers of antibody to EBV have been reported in lupus, suggesting that EBV may be a possible causative agent.73 Another example of potential antigenic mimicry is that between mycobacterial proteins and retinal autoantigens. Following systemic infection with Mycobacterium tuberculosis, or BCG therapy, an aggressive immune response in the eye can occur secondary to cross-reactivity of T cells that recognize both mycobacterial antigens as well as ocular proteins. Several homologous amino acid sequences were found between bacille Calmette-Guérin (BCG) proteins and retinal autoantigens that could explain the T-cell reactivity.74 Yet another interesting example is that of anti–Chlamydia pneumoniae antibodies, which have been found to be elevated in patients with age-related macular degeneration, suggesting a possible association.75
Interactions with Superantigens
Superantigens are immunostimulatory proteins produced by bacteria, such as group A streptococcus and staphylococcus, which bypass specificity to cross-link T-cell receptors with the MHC class II molecules of APCs in a nonrestricted manner.76,77 Studies suggest that superantigens exhibit varying binding affinity to different class II alleles, suggesting that particular HLA alleles create susceptibility to greater disease severity. For example, the class II allele HLA-DQA1*0, has shown preferential presentation of group A streptococcal proteins, inducing a high proliferative response.76,77
Presentation of Disease-Inducing Peptides
MHC class I and II molecules control the structure of the peptide-binding groove, which dictates the shape of the peptides that will fit.40 It has been hypothesized that certain disease-inducing peptides contain allele-specific motifs that allow them to be presented uniquely by HLA molecules encoded by the susceptibility genes.78 Additionally, aberrant forms of HLA molecules encoded by susceptibility genes, such as the heavy chain homodimer form of HLA B27, could uniquely present disease-inducing peptides.79 In the case of acute anterior uveitis (AAU), a possible link to the association with HLA-B27 includes molecular mimicry with Gram-negative bacteria Yersinia, Klebsiella, or Chlamydia and a deviant antigen presentation of the HLA-B27 antigen or even HLA class II presentation of a related B27 peptide.80 Furthermore, the most abundant naturally processed peptides from the HLA class II molecules are those derived from other HLA molecules. It is possible that certain HLA class II restricted T lymphocytes, stimulated by bacterial infection, may have a cross-reactive specificity for an HLA-derived peptide.
Aberrant T-Cell Receptor Development
An individual’s T-cell repertoire is a result of both positive and negative selection within the thymus. Disease-causing T cells, once selected by disease-associated HLA gene products, can evade the normal protective mechanisms of central tolerance. Once in the periphery, these T-cells could inappropriately recognize and respond to self peptides. One possible mechanism by which this can occur is by defective thymic selection, through a proposed “altered avidity model”.81,82 This model proposes that a weak interaction between peptide and HLA is overcompensated by a stronger interaction by the T-cell receptor, which promotes T-cell survival. T cells that become part of the peripheral repertoire as a result of overcompensation are more likely to be autoreactive.83 At this time, however, this mechanism does not seem to have a demonstrable role in eye diseases.40,63
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


