Chapter 21 The lacrimal system
Introduction
Lacrimal problems in children usually relate to underproduction of tears, causing dry eyes, which is rare but potentially sight-threatening, or reduced drainage of tears, which is much more common but less serious (Box 21.1).
Lacrimal gland
Embryology
The lacrimal gland develops from ectoderm that is supported by mesodermal connective tissue. It continues to grow 3–4 years after birth. Basal tearing is present in infants from birth, and reflex tearing begins at any time from birth to several months of age.1
Congenital abnormalities
Congenital absence is rare, usually occurring in conditions with reduced conjunctiva: anophthalmos, cryptophthalmos, and the lacrimo-auriculo-dento-digital (LADD) syndrome. Anomalous lacrimal ductules that secrete tears on to the skin rather than the conjunctival sac may be found near the lacrimal gland, around the lateral canthus, or in the preauricular region. These are rare but may require dissection and excision.2
Crocodile tears (see Chapter 100) occur from congenital aberrant innervation between the fifth and seventh cranial nerves causing tearing with chewing or sucking.3
Dry eyes in children
Congenital causes
Congenital alacrima is rare. It may be due to absence of the lacrimal gland or to it being ectopic in the orbit. Alacrima may be associated with systemic conditions such as the Riley-Day syndrome (familial dysautonomia), anhydrotic ectodermal dysplasia, and Allgrove’s syndrome (familial alacrima, achalasia of the cardia, and adrenal deficiency) (see Chapter 98).
Acquired causes
Acquired tear deficiency may be due to pathology of the lacrimal gland, causing failure of tear production, or to conjunctival damage (see Chapter 31), leading to ductule obliteration. The lacrimal gland may be damaged by Epstein-Barr infection, as the result of HIV infection, or in patients with bone marrow transplantation (often associated with graft-versus-host disease). The conjunctiva may be affected by injury (burns), infection, the sequelae of trachoma, Stevens-Johnson syndrome, or toxic epidermal necrolysis.
Sjögren’s syndrome is rare in children. It can be a primary autoimmune event or associated with rheumatoid arthritis or systemic lupus erythematosus. Children with Sjögren’s syndrome often have lacrimal gland enlargement. They may have recurrent parotid gland swelling and salivary gland involvement. Sjögren’s syndrome should be considered in any child with recurrent parotiditis, keratoconjunctivitis sicca, and early tooth decay due to xerostomia.4
Lacrimal tumors (see Chapter 26)
Lacrimal tumors are very rare in children. Pseudotumor causing painful swelling may affect the lacrimal gland.5 Malignant epithelial tumors, including mixed cell adenocystic and other carcinomas, have been recorded in childhood.6
Lacrimal gland enlargement is also found in conditions such as sarcoidosis or leukemia. Prolapse of the lacrimal gland, which is commonly bilateral, may present as a subconjunctival mass in the upper outer fornix. This may occur with craniofacial anomalies (see Chapter 28) due to reduced orbital volume and increased orbital pressure.
The lacrimal drainage system
Embryology
The lacrimal outflow system develops between the maxilla and the lateral nasal process from surface ectoderm. By the end of the first trimester this tissue begins to canalize. The puncta usually open with the eyelids during the sixth month of gestation. The nasolacrimal duct opens into the inferior meatus of the nose just before or after term birth. There may be a failure of this canalization process at any part of the system, but this is most frequent at the lower end.7
Congenital abnormalities
Common abnormalities, include narrowing (stenosis), blockage (atresia), complete absence (agenesis), or duplication (accessory channels) of any part of the system. A membranous obstruction at the distal end of the nasolacrimal duct is the commonest abnormality, causing congenital nasolacrimal duct obstruction.7 Obstruction at other sites is rare, but more relevant in older children.
Children with craniofacial abnormalities (see Chapter 28), particularly clefting syndromes, have complex anomalies of the lacrimal outflow system that may involve large areas being either blocked or absent.
Congenital dacryocystocele
A dacryocystocele is a congenital swelling located at the medial canthus due to trapped fluid inside the lacrimal sac and nasolacrimal duct.8 The fluid is unable to escape from either the upper or lower end of the drainage system as both are blocked. This presents as a tense, blue, non-pulsatile swelling below the medial canthus. It is evident at, or shortly after, birth (Fig. 21.1A). The inferior end of the dacryocystocele projects into the nose (Fig. 21.1B) and in some cases may be responsible for breathing difficulties.9 If respiratory compromise occurs, urgent treatment is required.
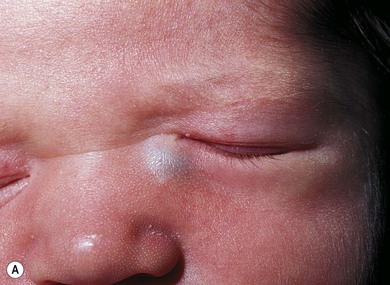
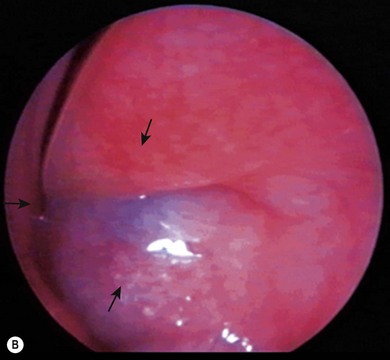
Congenital dacryocystocele must be differentiated from a meningoencephalocele, a meningocele, a mid-line nasal dermoid cyst (see Chapter 29), or a capillary hemangioma (see Chapter 20). An MRI scan is helpful in identifying the dilated sac and nasolacrimal duct and excluding other pathology. Routine imaging is unnecessary; the diagnosis is usually made clinically.
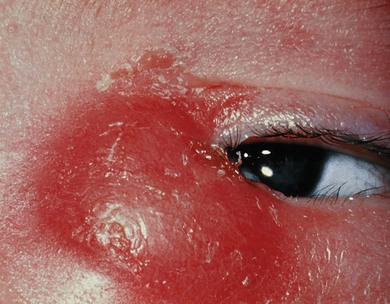
Treatment of a dacryocystocele involves observation during the first 2 weeks of life, during which time most spontaneously improve. If it has not settled by this stage, if acute dacryocystitis (Fig. 21.2) or respiratory difficulties develop, then endoscopic drainage of the dacryocystocele into the nose is indicated and the nasal mucosa over the dacryocystocele excised. If acute dacryocystitis has intervened, intravenous antibiotics should be given prior to surgery.
Congenital nasolacrimal duct obstruction
Congenital nasolacrimal duct obstruction represents a delay in maturation of the lacrimal system where it enters the nose, resulting in a persistent membranous obstruction at the valve of Hasner. The diagnosis is made on a history of a watery eye that has been present from the first few weeks of birth. This is usually unilateral but may be bilateral. If so, it is commonly asymmetrical. Some children develop a mucopurulent discharge that may be constant or intermittent. The eye remains “white” without evidence of active infection, although conjunctivitis may complicate the condition. The child is well with no evidence of irritation or photophobia. The skin around the eye may become red and excoriated. Although usually an isolated abnormality, congenital nasolacrimal duct obstruction may be more frequent in certain conditions, such as EEC syndrome (ectrodactyly, ectodermal dysplasia, clefting; Fig. 21.3) branchio-oculo facial syndrome,, craniometaphyseal or craniodiaphysial dysplasia, Down’s syndrome,, LADD syndrome, and the CHARGE association (Table 21.1).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


