The Eye in Skin and Mucous Membrane Disorders
C. Stephen Foster
Skin and eye are closely connected through at least three avenues:
Skin and mucous membranes and corneal epithelium are derived from common embryologic ancestry, with the surface epithelium deriving from surface ectoderm and the subepithelial connective tissues and vascular components deriving from mesoderm.
Juxtaposition between surface of the eye and edge of the eyelids can result in “innocent bystander” ocular damage when a particular disorder is affecting only the skin but is affecting the skin of the eyelids up to the lid margin.
Certain immune disorders, such as cicatricial pemphigoid, eczema, xeroderma pigmentosum, and Vogt-Koyanagi-Harada syndrome, to name a few, may result in an autoimmune attack of both ocular structures, such as conjunctiva, and skin.
The importance of these shared properties between the eye and skin is relatively underappreciated by ophthalmologists, and it is the purpose of this chapter to emphasize this connection and its importance to the ophthalmologist, particularly the ability to diagnose systemic disease on the basis of its ocular manifestations. In addition, it is important for the ophthalmologist to recognize that proper care of the eye will categorically require concomitant care of the skin and treatment of the underlying systemic disease.
The diseases chosen for discussion here are primarily the more serious, potentially blinding skin disorders that can affect the eye, and some of the more common disorders with which the ophthalmologist will have contact on multiple occasions (e.g., rosacea). The reader is referred to other excellent resources for additional reading.1,2
Ocular abnormalities associated with disease of the skin and mucous membranes may involve any part of the eye or its adnexae. In this discussion, the dermatologic conditions will be grouped in broad categories, and the eye manifestations will be discussed with each disease as follows:
Bullous-Vesicular Disorders
Pemphigus
Cicatricial pemphigoid
Bullous pemphigoid
Dermatitis herpetiformis
Erythema multiforme
Toxic epidermal necrolysis
Epidermolysis bullosa
Hydroa vacciniforme
Acrodermatitis enteropathica
Pigmentary Disorders and Diseases Precipitated by Light
Incontinentia pigmenti
Xeroderma pigmentosum
Chédiak-Higashi syndrome
Nevus of Ota
Hyperkeratotic Disorders
Ichthyosis
Psoriasis
Atopic dermatitis
Disorders of Skin Elasticity
Ehlers-Danlos syndrome
Pseudoxanthoma elasticum
Disorders Involving Neoplasia
Basal cell nevus syndrome
Juvenile xanthogranuloma
Hemangioma
Miscellaneous Skin Disorders
Acne rosacea
Anhidrotic ectodermal dysplasia
Malignant atrophic papulosis
Focal dermal hypoplasia syndrome
BULLOUS-VESICULAR DISORDERS
PEMPHIGUS
Description
Pemphigus (from the Greek meaning puff or blister) comprises a group of autoimmune vesiculobullous eruptions of the skin and mucous membranes that are seen primarily in middle-aged people, with both sexes being affected equally.3 Involvement of mucosae may occur as the initial manifestation in as many as 25% of cases, and it occurs at some point during the course of disease in 95% of cases.4 Pemphigus vulgaris and its variant, pemphigus vegetans, constitute a more acute form of the disease in which the earliest lesions appear in the oral mucosa and are then followed by a bullous eruption of the skin over the trunk and extremities within 4 to 12 months (Fig. 1). This form of pemphigus was almost uniformly fatal prior to the use of systemic corticosteroid therapy. The mortality rate was reduced to approximately 40% by the use of steroids and has been further reduced to 5% by the addition of immunosuppressive chemotherapeutic regimens for the most difficult cases. Pemphigus foliaceus and its variant, pemphigus erythematosus, follow a more chronic but benign course. Mucosal lesions are unusual in this form, which is primarily an exfoliative dermatitis.3
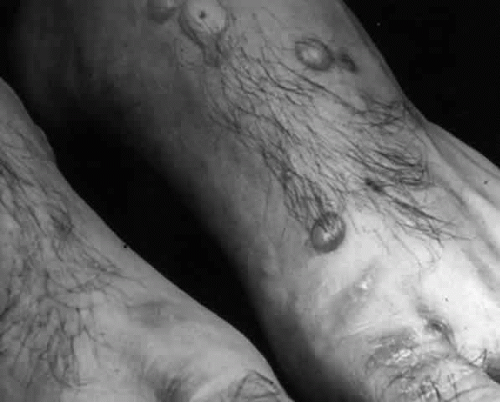 Fig. 1. Pemphigus vulgaris. Bullae are flaccid, particularly when they enlarge. (Courtesy of Dr. Howard Bader, Massachusetts General Hospital) |
The characteristic histologic feature of pemphigus is acantholysis or loss of interepidermal cell bridges. This leads to bulla formation within the epidermis. The epidermis may be readily dislodged by lateral sliding finger pressure, the Nikolsky sign, a clinical feature shared with toxic epidermal necrolysis and epidermolysis bullosa.4
In pemphigus vulgaris, the bullae form above the basal cell layer; in the foliaceus and erythematosus forms of pemphigus, such changes occur just below the stratum corneum. Marked acanthosis, with formation of intraepidermal eosinophilic abscesses, is present in pemphigus vegetans. These changes are found in biopsy specimens of both skin and mucous membranes.5
Epidemiology
Pemphigus vulgaris is by far the most common form of pemphigus, occurring predominantly in people of Jewish or Mediterranean origin, but it is reported in all races and ethnic groups, with no sex predilection.
Pathogenesis
Antibodies to intercellular components of epidermis have been demonstrated in the sera of patients with pemphigus.6 Tissue-fixed immunoglobulins are present in the interepidermal cell spaces, and a large percentage of these patients have circulating antibodies that react with an intercellular antigen present in many stratified squamous epithelia.5 These intercellular substance-reactive antibodies are typically IgG, are complement fixing, are pathogenic to tissue culture preparations of epidermal cells,7 and produce blistering skin lesions, identical to pemphigus vulgaris lesions, in mice injected intraperitoneally with the antibodies.8 The relevant antigen in the intercellular substance is a cadherin.9 The disease develops in the genetically susceptible individual, probably after he or she is exposed at some point in life to an environmental trigger. The disease is especially prevalent in Ashkenazi Jews, and 92% of these patients express the major histocompatibility complex class II alleles HLA-DR4, DQw8.10 It is presumed that because the disease is attributable to the presence of an antibody to an intraepidermal intercellular cement substance, the class II susceptibility gene controls the production of the antibody as a dominantly expressed immune response gene.
The intercellular cement substance against which the autoantibody is directed, pemphigus vulgaris antigen (PVA), is a novel cadherin, limited in tissue distribution to stratified squamous epithelia, with significant homology with the other known cell adhesion molecules within the cadherin family (E-cadherin, N-cadherin, P-cadherin, L-CAM, and DGI). PVA like DGI binds to plakoglobin, and with DGI forms a subfamily of cadherins; the other cadherins bind to catenions and to CAP102. DGI is the target antigen in pemphigus foliaceus and is present in all desmosome-containing tissues.9
Diagnosis
Pemphigus can be differentiated from bullous pemphigoid and from cicatricial pemphigoid on histologic and immunologic bases. Whereas the bullae in pemphigus are intraepidermal, those occurring in bullous pemphigoid and in cicatricial pemphigoid are subepidermal, between epidermis and dermis, or between mucosa and submucosa. Acantholysis is not present in the latter two conditions.5 In bullous pemphigoid and in cicatricial pemphigoid there are tissue-fixed immunoglobulins in the subepidermal basement membrane zone (Fig. 2).11 This is in contrast to the interepidermal fixation of antibodies (Fig. 3) seen in pemphigus (Table 1).
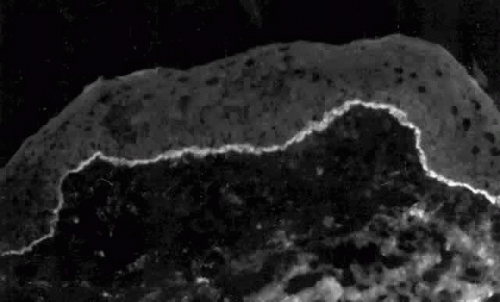 Fig. 2. Fluorescence microscopy of conjunctiva from a patient with cicatricial pemphigoid affecting the eye. The antibody used was a fluorescein-conjugated antibody directed against human IgG. Note the bright, continuous, linear deposition of IgG at the epithelial basement membrane zone, a finding virtually diagnostic of cicatricial pemphigoid. |
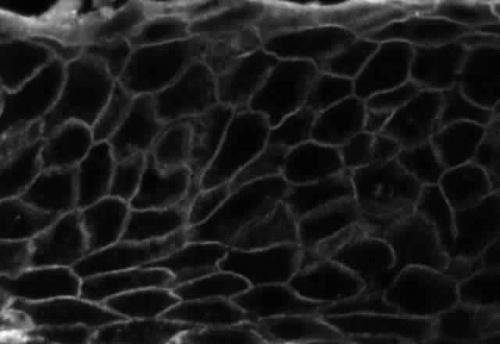 Fig. 3. Immunofluorescence microscopy of conjunctiva from a patient with pemphigus vulgaris. The antibody is a fluorescein-conjugated antibody directed against human IgG. Note the bright staining of the intercellular cement, brightly outlining the borders of all of the epithelial cells. |
TABLE 1. Histologic and Immunologic Features of the Blistering Dermatoses | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Ocular Manifestations
Ocular complications in pemphigus are rare,5 but ocular manifestations of pemphigus on lids, conjunctiva, cornea, lens, and iris have been reported. Conjunctival bullae are infrequently seen in pemphigus vulgaris. More typically, a catarrhal or purulent conjunctivitis develops.3 The ocular features are similar in pemphigus foliaceus and are usually confined to the palpebral conjunctiva, with erythema, edema, and purulent discharge.5,12 Pemphigus foliaceus may also produce entropion and trichiasis of both lids with resultant corneal damage. Lens opacities have been described in 5% of patients with a form of pemphigus foliaceus that is endemic to Brazil. Some of these patients also exhibit red, nodular iris lesions, the exact nature of which is not known.12
The conjunctival involvement in pemphigus may lead to symblepharon as a result of the formation and rupture of small vesicles. This does not, however, lead to progressive scarring and blindness, as is so often the case in cicatricial pemphigoid (Fig. 4).13
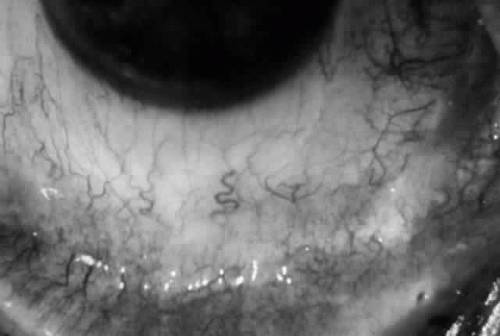 Fig. 4. Eye of a patient with ocular involvement in pemphigus vulgaris. Note the subepithelial fibrosis with fornix foreshortening and symblepharon formation. The degree of scarring stabilized since the patient’s pemphigus vulgaris was brought under control 5 years before photograph was taken. |
Therapy
Acute pemphigus vulgaris requires high-dose systemic corticosteroid therapy (100–200 mg prednisone/day), with gradual reduction of steroid dose with cessation of new blister formation.14 Infection is the leading cause of death in patients with pemphigus vulgaris; great vigilance for and vigorous treatment of any infection is obviously indicated. Immunosuppressive chemotherapy with methotrexate (15–25 mg once a week), azathioprine (2–3 mg/kg/day), or cyclophosphamide (2–3 mg/kg/day), used for long-term control of the disease, should be started once the risk of infection has diminished, with the usual caveats vis-a-vis monitoring for and managing drug toxicity.15,16
CICATRICIAL PEMPHIGOID
Definition
Cicatricial pemphigoid (CP) is a chronic cicatrizing autoimmune disease of the mucous membranes and skin. It is said to have an average age of onset of 65 years, but this figure under-reports the true epidemiologic features of this disease, because the cases are usually not in their earliest stages.17 Females are affected two to three times as frequently as males. Conjunctival involvement may occur as early as 10 years before other mucosal or skin lesions develop, or it may occur as late as 20 years after the onset of other lesions; the disease may be limited to the conjunctiva (Fig. 5). The scarring Brusting-Perry dermatitis occurs in approximately 25% of cases (Fig. 6), and cicatrizing conjunctivitis develops in 70% to 75%.17,18 Involvement of other mucosa may lead to scarring of the soft palate and oral and nasal mucosa, and esophageal, urethral, vaginal, and anal strictures may develop. Laryngeal involvement may cause pain and hoarseness,19 and the esophageal scarring can be lethal, because asphyxiation can result from a food bolus that is lodged during attempted swallowing by a patient with dysphagia from esophageal strictures that have been neglected by patient and physician alike.
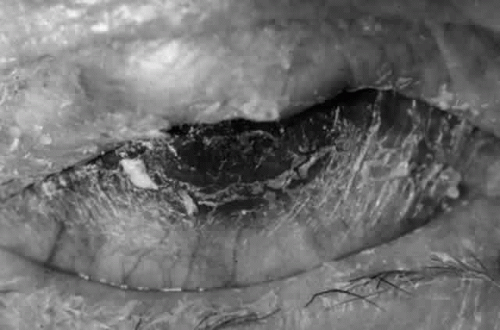 Fig. 5. Eye of a patient with cicatricial pemphigoid, biopsy-proven, without extraocular manifestations. The disease has progressed to end stage and blindness. Note the total “leatherization” of the eye, with complete adherence of the eyelids to the globe. |
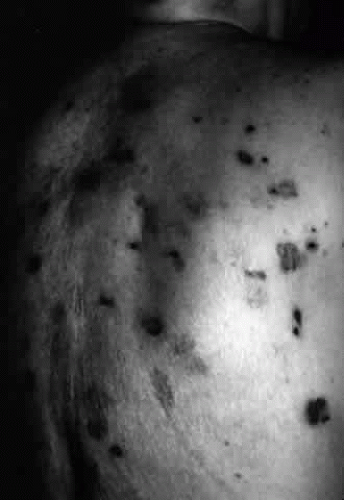 Fig. 6. Brusting-Perry dermatitis of cicatricial pemphigoid affecting skin. Note the scarring characteristic of this dermatitis of cicatricial pemphigoid, a characteristic quite different from that of bullous pemphigoid. |
Histologically, the conjunctival lesions show submucosal scarring,20 chronic inflammation, perivasculitis, and squamous metaplasia of the epithelium, with loss of goblet cells; mast cell participation in the inflammation is surprisingly great.
Tissue-fixed immunoglobulins and complement components are present in the epithelial basement membrane zone (BMZ) in patients with cicatricial pemphigoid20,21; indeed, such immunoreactant deposition is the sine qua non for definitive establishment of the diagnosis.20 Circulating antibodies to the basement membrane of conjunctiva are found in all of these patients if ultrasensitive radioimmunoassay techniques are employed,22 and circulating antibodies to conjunctival epithelium have been found (see Table 1).21,23,24 Circulating antinuclear antibodies (ANA) have also been demonstrated in patients with cicatricial pemphigoid.20,25
Epidemiology
Cicatricial pemphigoid is probably not quite as rare as published incidence figures indicate. Bittelheim26 estimated 1 in 15,000 ophthalmic patients; Hardy and Lamb27 and others28,29 estimated an incidence of 1 in 20,000 patients. Smith and colleagues30 estimated 1 in 46,000. Bedell31 estimated 1 in 8000, and Lever and Talbott32 stated frankly that it is impossible to make an accurate estimate from available data; they guess that the incidence of CP is somewhere between 1 in 12,000 and 1 in 60,000 ophthalmic patients. But these figures estimate the incidence of relatively advanced CP. Diagnosis of this disorder in its early stages is difficult,28 and most cases are not recognized as CP until they reach what we could categorize as stage III disease. There are, therefore, patients with stage I and stage II CP uncounted in the epidemiologic estimates.
Data on the average age of CP patients are also somewhat distorted by subtleties of the diagnostic signs. CP is said in many publications to be a disease of older people, with the average age given as 60)33 or 70)28 years. Although reports of CP in children may in fact represent cases of localized erythema multiforme, we certainly now recognize that CP can begin at least as early as the third decade of life.
Most reports describe a slight but clear female predilection. Thus, Klauder and Cowan34 reported 8 females and 3 males, Lever35 reported 3 females for every male, and Hardy and colleagues36 reported 52 females and 29 males with CP; 62 of these patients had ocular involvement, but Hardy and associates36 did not report the sex distribution for this subset of patients. No racial or geographic predilection is reported.36
Pathogenesis
Ocular cicatricial pemphigoid (OCP) is clearly an autoimmune disease with a genetic predisposition requirement and probably a “second-hit” environmental requirement to trigger the onset of the disease. We first reported an increase in the frequency of the HLA-DR4 and HLA-DQw3 alleles in OCP patients.37 Subsequently, in patient and family studies employing restriction fragment length polymorphism analysis of the DQw3 haplotypes, it was determined that the gene that provides enhanced susceptibility to OCP development is the HLA-DQw7 gene.38
The second-hit environmental trigger that stimulates the genetically susceptible individual to develop OCP may be microbial (we suspect that this is so for idiopathic OCP) or may be chemical, as in the case of so-called drug-induced or pseudo-OCP, which develops in some individuals exposed to practolol39 or to a limited variety of ocular medications.40,41,42,43 We know that patients with pseudo-OCP carry the same susceptibility gene (HLA-DQw7),44 but we further know that the autoantigens in these two forms of OCP are different. We have identified a 205-kd protein molecule in the basement membrane zone of conjunctiva and epidermis as the relevant target antigen in idiopathic OCP,45 and we have found that sera from patients with pseudo-OCP bind to 97- and 290-kd proteins in conjunctiva and epidermis lysates, and to 45-, 150-, 290-, and 400-kd proteins in dermal lysates.46 None of these autoantigen protein targets is absorbed from the tissue lysates by sera from patients with bullous pemphigoid, pemphigus vulgaris, or OCP, indicating that the autoantigens in OCP, pseudo-OCP, and bullous pemphigoid are distinct from each other.
The autoantibodies that develop in patients with OCP22 can be detected in all OCP patients when the disease is active, and the autoantibodies are probably pathogenic, just as the autoantibodies in patients with pemphigus are.7,8 The binding of the autoantibody to the autoantigen at the epithelial basement membrane then sets in motion a complex drama in which an impressive cast of characters participates.20 CD4 (helper) T lymphocytes far outnumber CD8 (suppressor) T cells in the inflammatory cell population that develops in the substantia propria of the conjunctiva; plasma cells, histiocytes, and mast cells are present in very large numbers, and a panoply of cytokines is elaborated from these cells, the effects of which are only now being dissected by molecular biologic techniques in our laboratory. However, the result of this complex drama is corneal epithelial damage from inflammatory cytokines and from the xerosis, meibomian gland dysfunction, and trichitic trauma developing as a consequence of cytokine-induced conjunctival fibroblast proliferation and activation, with resultant subepithelial fibrosis.
Diagnosis
The diagnosis of OCP is extremely important, given the natural history of the disease, the effectiveness but potential toxicity of therapy, and the potential confusion in differentiation from other causes of chronic cicatrizing conjunctivitis (Table 2). The clinical diagnosis requires immunohistochemical confirmation prior to institution of therapy20; the diagnosis is confirmed by the demonstration of one or more immunoreactants at the epithelial basement membrane zone (see Fig. 2).
TABLE 2. Causes of Cicatrizing Conjunctivitis | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Ocular Manifestations
Clinically, the ocular disease in cicatricial pemphigoid (OCP) may present unilaterally in the form of a chronic, recurrent catarrhal conjunctivitis, but it eventually becomes bilateral. Subepithelial fibrosis is characteristic of stage 1 of OCP (Fig. 7). Stage 2 shows fornix foreshortening (Fig. 8), and symblepharon formation is the hallmark of stage 3 (Fig. 9). Stage 4, end-stage disease, is characterized by ankyloblepharon and surface keratinization (Fig. 10). Obstruction of the lacrimal ductules and meibomian gland ducts eventually produces an unstable tear film and progressive sicca syndrome, but it is to be emphasized that OCP is not a dry-eye syndrome until late in the disease course.20 Trichiasis and entropion occur because of the subepithelial fibrosis, with eventual keratopathy, corneal neovascularization, and corneal ulceration and scarring.20
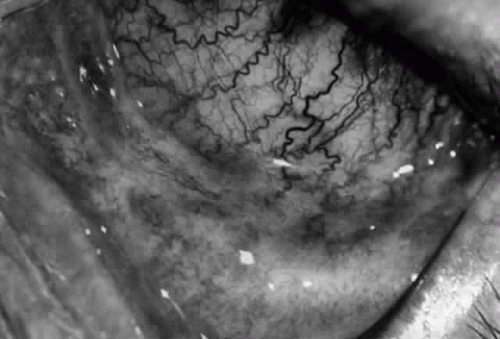 Fig. 7. Stage 1 cicatricial pemphigoid, with cicatrizing conjunctivitis, and fine striae-type areas of subepithelial fibrosis, but without evidence of shrinkage of the conjunctiva. |
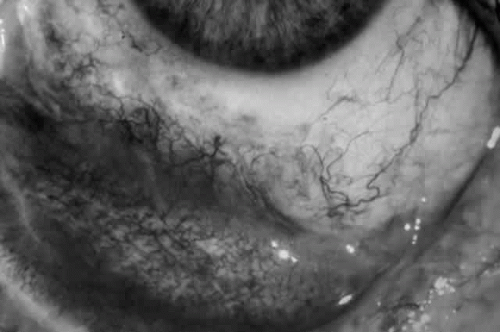 Fig. 8. Stage 2 cicatricial pemphigoid, with fornix foreshortening and subepithelial fibrosis without frank symblepharon formation. |
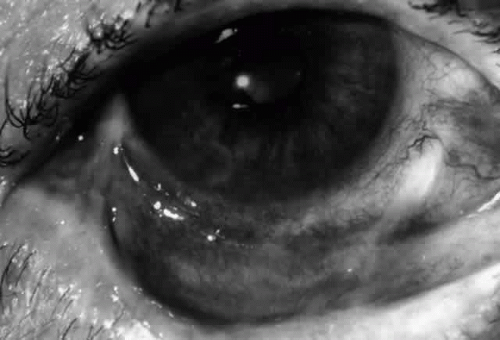 Fig. 9. Stage 3 of an eye affected by cicatricial pemphigoid. The conjunctival “shrinkage” continued and a frank symblepharon developed. |
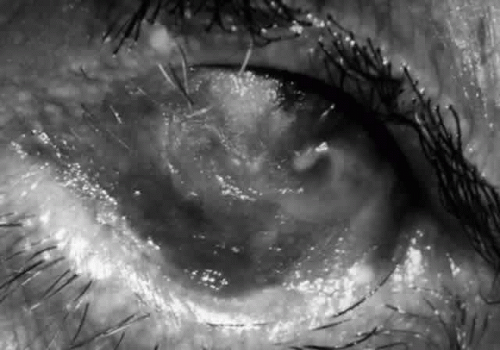 Fig. 10. Stage 4 cicatricial pemphigoid. Progressive shrinkage of the conjunctiva resulted in extreme trichiasis and distichiasis and keratopathy, with compromise of meibomian ductules and lacrimal ductules and the production of a totally dry eye. |
In a series reported from the Mayo Clinic, 21 of 81 patients with cicatricial pemphigoid were blind at the time of the report; 17 of these patients were blind in both eyes. Sixty-two of the 81 patients had ocular involvement; approximately 88% of the 62 patients had the ocular disease bilaterally.18
Therapy
Medical treatment of the ocular complications of cicatricial pemphigoid was once difficult, protracted, and not very effective. Herron reported on the management of a case with ocular, skin, and mucosal involvement of the pharynx and larynx. Oral triamcinolone was used, along with injections of the drug into the areas of symblepharon. The skin and mucosal lesions responded in 10 weeks, but the ocular disease did not.47
CURRENT STATUS OF MEDICAL THERAPY PROGRAM FOR CP.
Our current approach to medical treatment of patients with active CP20 may be summarized as follows:
Sicca syndrome is treated aggressively. We favor ointment lubricants without preservatives (e.g., Akwa Tears, Optilube, or Duolube ointments) applied at a frequency sufficient to maintain adequate surface lubrication and protection (e.g., every 2>4 hours and at bedtime). Artificial tears without preservatives (e.g., Refresh or Unisol drops) are employed as needed between ointment applications.d—Topical retinoid (0.01% tretinoin) ointment is used once or twice daily in one eye; we compare objective and subjective responses to the vitamin A ointment and continue treatment beyond 4 weeks of therapy only if we and/or the patient concludes that there is a distinct therapeutic benefit to this agent. Fewer than 30% of our CP patients have shown an apparent benefit from topical retinoids. We strongly caution against inappropriate enthusiasm for the efficacy of this component of the therapeutic plan. We observed the unfortunate results of such inappropriate preoccupation with topical therapy for this systemic disease in three patients who are now functionally blind because the disease progressed while they were on topical retinoids without concomitant appropriate systemic therapy.
Chronic blepharitis and meibomianitis are treated with vigorous lid hygiene and oral doxycycline, 100 mg once or twice daily. Manual expression of plugged meibomian ducts is performed at the time of every evaluation. Microbial colonization of the lids and meibomian glands by any organism, including Staphylococcus, is treated with appropriate topical and/or systemic antimicrobial agents. We have discovered two cases of chronic Candida infestation and one of Nocardia.
Chemotherapy for cases that are extremely active (4+) and rapidly progressive is begun with prednisone 1 mg/kg and cyclophosphamide 2 mg/kg/day. The cyclophosphamide dose is adjusted according to therapeutic response, bone marrow response, and drug tolerance. Prednisone is gradually tapered, with a switch to alternate-day dosing by 8 to 12 weeks and subsequent tapering and discontinuation by 4 to 6 months. We presently maintain patients on chemotherapy for a minimum of 1 year.
Dapsone and prednisone are used for cases that are less active and cases that are not rapidly progressive. The initial daily dose of dapsone we employ is 1 mg/kg; the maximum dose ever employed is 200 mg/day. We emphasize that dapsone is not a benign drug. We follow our patients who are on dapsone as often and as carefully as we do those who are on cyclophosphamide. Patients who are not glucose-6-phosphate dehydrogenase deficient, as determined by the screening test, may still hemolyze when treated with dapsone; therefore, the hemoglobin, hematocrit, and reticulocyte count, as well as the white blood count, are important monitoring parameters for these patients. We usually accept a low degree of hemolysis if dapsone is achieving the desired therapeutic response. However, reticulocytosis must adequately compensate for such hemolysis; a steadily falling hematocrit is unacceptable.
Approximately 70% of patients will respond well to dapsone. A large proportion of these (41% in our experience) will relapse within 6 months of cessation of dapsone therapy.48 We treat these patients with dapsone, methotrexate or azathioprine. Azathioprine, 2 mg/kg initial dose, is also our second-choice agent for the 30% of patients who do not respond adequately to dapsone. Adjustment of azathioprine dose is accomplished, as in the case of the other agents, through the judgment of the skilled chemotherapist, who, by virtue of training and experience, is an expert in the use of these agents.
Some authors have advocated surgical treatment in the early or “moist” phase of the disease: tarsectomy for correction of entropion, strip peritomy to provide an avascular barrier against corneal vascularization, superficial keratectomy for removal of corneal vascular and scar tissue, and fornix incision for release of symblepharon. All of these approaches not only will fail but in fact will make the disease worse if the patient is not adequately immunosuppressed prior to surgery. Mucous membrane grafting may be of benefit in cases of cicatricial entropion and lid marginal keratinization associated with this disease.49 Contact lenses and shells with concurrent use of lubricants may aid in the management of the chronic, dry phase of the disease with corneal involvement.50
BULLOUS PEMPHIGOID
Definition
Bullous pemphigoid (BP) is an autoimmune blistering dermatosis first recognized by Lever as a distinct entity in 1953.51 It clinically resembles, but is not identical to, pemphigus vulgaris (hence the name pemphigoid), and is one of four diseases on the spectrum of bullous pemphigoid, cicatricial pemphigoid, linear IgA disease, and herpes gestationis. The latter typically develops in pregnant and postpartum women, resolving within months and recurring with each pregnancy. Like pemphigus vulgaris, BP is characterized by skin bullae that may rupture; the disease can be fatal, usually as a result of sepsis.
Epidemiology
BP typically occurs in individuals of either sex between the ages of 50 and 80, without racial or geographic predilection. Its true prevalence and incidence are unknown.
Pathogenesis
A genetic predisposition to this disease, HLA-DQw7 accompanied by some (as yet unknown) environmental trigger, results in IgG autoantibodies that recognize and attach to 180-kd and 230- to 240-kd proteins associated with the hemidesmosomes and the lamina lucida of the epidermal basement membrane zone.52,53 The 230-kd autoantigen, or BPAG1, is a glycoprotein encoded on chromosome 6p,52 whereas the 180-kd autoantigen, or BPAG2, is a novel collagen encoded by a gene on the long arm of chromosome 10, at locus 10q24.3.53 These proteins or autoantigens are distinct from those recognized by the autoantibodies of cicatricial pemphigoid, pemphigus vulgaris, dermatitis herpetiformis, and epidermolysis bullosa.
The BP autoantibodies are complement fixing, and they are pathogenic. Fixation of the autoantibodies to the BP antigens, followed by complement fixation, results in complement split-product-mediated attraction of neutrophils and eosinophils, with release of their cytokines and production of tissue damage and blister formation.54 Mast cells also play a role in lesion development,55 and itch is a common symptom during acute phases of the disease. The disease is chronic, remitting, and recurrent when untreated, lasting from months to years, with little or no tendency for the healing skin lesions to scar. Death, however, may occur as a result of sepsis.
Diagnosis
The diagnosis of BP is suspected in the patient (usually 50–80 years old) with a blistering dermatosis with large tense (rather than flaccid) bullae (on a normal or an erythematous base) that has a predilection for intertriginous areas, palms, soles, and abdomen (Fig. 11). Nikolsky’s sign is negative. Mucosal lesions (blisters) may be present in approximately 30% of patients. The histopathologic characteristics of a biopsied skin lesion include the presence of subepidermal blister formation without acantholysis, with neutrophils, eosinophils, and lymphocytes in the blister cavity and a lymphocytic perivasculitis in the dermis. The hallmark immunopathologic feature of BP is the presence of IgG and the third component of complement (C3) at the dermal-epidermal (DE) junction and the presence of anti-BMZ antibodies, usually in high titer, in the serum of the patient. These can be detected by indirect immunofluorescence technique using guinea pig lip or monkey esophagus as substrate.
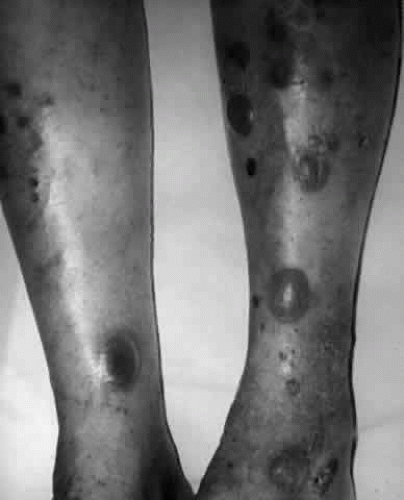 Fig. 11. Bullous pemphigoid. Note the tense bullae in this patient whose dermatitis is associated with bullous pemphigoid. This patient is in the erect position. If these were bullae of pemphigus vulgaris, they would “droop” because of their flaccidity. |
Ocular Manifestations
Ocular manifestations of BP are not as impressive (Fig. 12) as those of cicatricial pemphigoid affecting the conjunctiva; therefore, they may go undetected. A study of 36 patients with BP in England disclosed that some had fine striae of tarsal subepithelial fibrosis, and one had frank symblepharon formation.56 Seventy-three percent of those who had conjunctival biopsy performed showed immunoreactant deposition at the BMZ.57
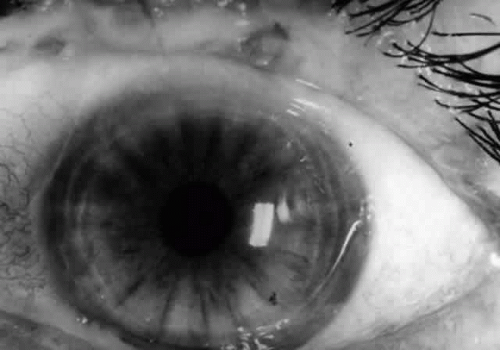 Fig. 12. Conjunctivitis and eyelid margin blepharitis in a patient with bullous pemphigoid. Note the small vesicle at the lid margin. |
DERMATITIS HERPETIFORMIS
Definition
Dermatitis herpetiformis (DH), first described by Duhring in 1884,60 is an autoimmune blistering dermatosis characterized by an intensely itching papulovesicular rash, especially on extensor surfaces, and associated (usually) with asymptomatic gluten-sensitive enteropathy.
Epidemiology
DH affects both sexes of any age, with peak incidence at ages 10 to 40 years. It is unclear how common this disease is.
Pathogenesis
Just as in cases of systemic lupus erythematosus, myasthenia gravis, celiac disease, and juvenile onset diabetes mellitus, dermatitis herpetiformis is associated with the HLA-B8, HLA-DR3, DQw2 phenotype. Such genetically predisposed individuals tend to make IgA autoantibodies directed against proteins in the dermal papillary tips, specifically in anchoring fibrils below the basal lamina.61 They may be provoked to make such antibodies through gastrointestinal contact with gluten, a protein in rye, barley, oats, and wheat.62 IgA and/or IgA-containing immune complexes deposit in the apices of the dermal papillae and activate complement via the alternative pathway, thereby attracting neutrophils, with resultant microabscess formation and the clinical appearance of the papulovesicular rash characteristic of DH.63
Diagnosis
Herpetiform (herpeslike) groupings of itchy papulovesicular eruptions on elbows, knees, shoulders, buttocks, and hands, preceded by an 8-hour itchy, burning prodrome, make the diagnosis nearly unmistakable (Fig. 13). The diagnosis is confirmed by the histopathologic findings, on lesion biopsy, of microabscesses in the apices of the dermal papillae, by the immunopathologic finding of granular IgA deposits in the same locus, and, most important, by the finding of granular IgA deposits in normal-appearing skin. The third component of complement (C3) is usually found along with IgA. Finally, the diagnosis is confirmed by the extremely rapid (within days) resolution of the lesions after starting dapsone therapy (see Treatment section).
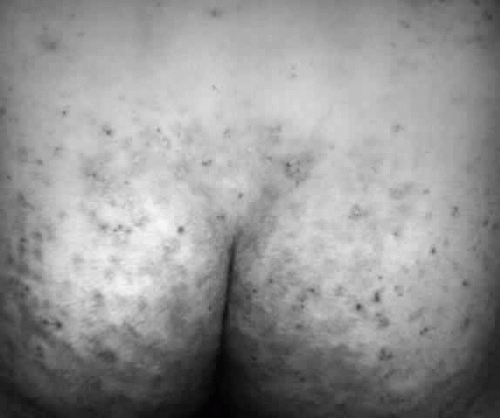 Fig. 13. Dermatitis herpetiformis. Note the grouped vesicular and small bullous arrangement of this dermatosis, which is intensely pruritic. |
Ocular Manifestations
Ocular manifestations of dermatitis herpetiformis are rare. Keratoconjunctivitis sicca, iritis, choroiditis, and papillitis have occurred in individual patients, but the causal relationship between dermatitis herpetiformis and the ocular problem is debatable.
Treatment
Dapsone revolutionized the care of patients with DH, with resolution of symptoms and new lesion development within 48 hours of initiation of therapy.64 The usual caveats regarding dapsone therapy vis-à-vis sulfa allergy and glucose-6-phosphate dehydrogenase deficiency must obviously be kept in mind. The dose of dapsone sufficient to induce remission, 100 to 300 mg per day, can usually be tapered to very low doses for maintenance of remission. Equally important, however, is the strict adherence to a gluten-free diet if one is to eliminate the dependence on medical therapy.65
ERYTHEMA MULTIFORME
Definition
Erythema multiforme (EM) is an acute inflammatory disease affecting primarily skin and mucous membranes.66 Major and minor variants exist, and it is the former, also known as Stevens-Johnson syndrome (SJS), that is of most concern to ophthalmologists.67 Prodromal symptoms of SJS include general malaise, myalgias, and fever.68 Skin lesions develop by 10 days to 4 weeks after the onset of the prodrome; they tend to be annular in shape (“iris lesions”) but may be maculopapular or bullous. Most commonly, involvement is limited to the skin of the dorsum of the hands and feet and the palms and soles. Later, the limbs, trunk, face, and neck may become involved (Fig. 14). EM major or SJS may be fatal, either from the systemic, visceral involvement of the disease or from complications arising from the extensive exfoliation of skin, producing consequences similar to those seen in patients with third-degree burns. The mortality rate is reported to be between 5% and 15% of cases, and 20% of cases recur.66
 Fig. 14. Erythema multiforme major. This patient has oral mucous membrane involvement in addition to the classic target dermatitis lesions on his arms. |
Epidemiology
Pathogenesis
Histologically, the skin lesions exhibit diffuse inflammatory infiltration and edema of the dermis. When bullae are present, they are subepidermal in location and without acantholysis.66
The exact pathogenetic mechanism of this disease is unknown; autoimmune, infective, and allergic factors have all been considered possible causes. Most evidence now favors a hypersensitivity to microbes and drugs.70 Erythema multiforme has been associated with various bacterial (especially Mycoplasma pneumoniae), viral (especially herpes simplex virus), mycotic, and protozoan infections, with drugs and vaccines, with allergic contactants, with collagen disorders, and with internal malignancies. Some of the incriminated precipitating drugs include sulfonamides, tetracyclines, penicillin, bromides, iodides, salicylates, phenytoin sodium, barbiturates, phenylbutazone and other nonsteroidal antiinflammatory drugs, cortisone, and vaccines against polio, smallpox, influenza, diphtheria, and tetanus. The development of or exacerbation of Stevens-Johnson syndrome has been reported after ophthalmic application of proparacaine,71 sulfonamide,72 tropicamide,73 and scopolamine.73
Herpes simplex virus (HSV) is a vastly underappreciated cause of EM,74 with 15% to 63% of cases associated with this virus.75,76 Recurrent EM is especially associated with recurrent HSV infection,77 and herpes simplex virus has been isolated from the skin lesions of erythema multiforme.78
The results of our research,79 as well as that of others,80,81,82,83,84,85,86,87 suggest that episodic showers of circulating immune complexes may result in immune complex deposition in previously damaged, predisposed vessels, with resultant immune complex vasculitis/perivasculitis with lymphocyte and neutrophil participation in the vessel damage. Leakage of plasma and cells may progress to subepidermal blister formation and to necrosis of the epidermis over the blister.66
Diagnosis
The diagnosis of SJS is not difficult, given the dramatic characteristic skin and mucous membrane lesions.
Ocular Manifestations
Ocular involvement is common in Stevens-Johnson syndrome, occurring in more than half of patients with this disease.70 The acute ocular manifestations vary from mild mucopurulent conjunctivitis to severe, pseudomembranous conjunctivitis leading to symblepharon, lid scarring, keratoconjunctivitis sicca, corneal ulceration, corneal scarring, and neovascularization. The chronic consequences of SJS depend on the degree of subepithelial fibrosis and tarsal conjunctival keratinization that occurs as a result of the acute exanthem. The former (fibrosis) results in variable degrees of meibomian duct and lacrimal ductule obstruction (with resultant deficient or unstable tear film problems), trichiasis, distichiasis (with resultant chronic abrasive trauma to the cornea), and cicatricial entropion; tarsal conjunctival keratinization results in a chronic sandpapering trauma effect on the cornea with each blink. The combined effects of these external aggravants produce chronic keratopathy, with eventual corneal scarring, neovascularization, or ulceration (Fig. 15).
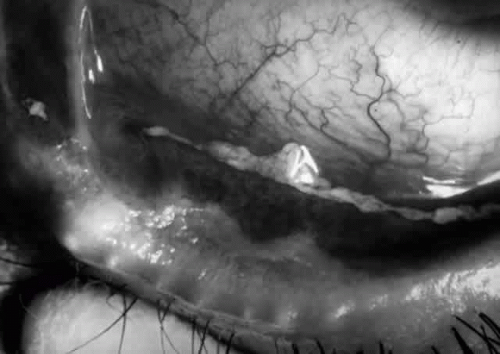 Fig. 15. Eye of a patient with Stevens-Johnson syndrome. Note the conjunctivitis, the subepithelial fibrosis, the symblepharon formation, and the zone of variable width, posterior to the gray line of the eyelid that is obviously keratinized. |
One other chronic, rare ocular manifestation of SJS may develop: recurrent, immunologically mediated conjunctivitis. Just as such recurrent episodes may occur in skin87 and in oral mucosa,88 so too may they occur in conjunctiva.79 The diagnosis of this phenomenon is not easy and depends on eradication of all external aggravants, eliminating these as confounding variables, and then demonstration, by immunohistochemical techniques employed on a biopsy specimen from the inflamed conjunctiva, of a lymphocytic microangiopathy with IgA deposition in the vessels. Herpes simplex virus is one inciting agent for recurrent SJS, and this possibility should be carefully investigated.
Therapy
Therapy of the acute phase of the ocular disease includes use of topical steroids as well as topical antibiotics applied frequently to combat superinfection, especially that from Staphylococcus aureus and (if the patient is hospitalized in a burn unit) Pseudomonas species. Lubricants should be generously employed, especially if lid involvement has resulted in even mild lagophthalmos. If anterior uveitis is present, mydriatic therapy is appropriate. Howard has pointed out that the pseudomembranes should not be stripped, because doing so leads to bleeding and increased scarring.89 Lid-globe adhesions should be lysed twice daily to maintain the fornices. I use my scleral depressor for this purpose after cleaning it with 70% isopropyl alcohol, having found no advantage to use of a glass rod. In an effort to prevent corneal scarring, Arstikaitis has advocated early tarsorrhaphy before deep stromal vessels invade the cornea.90 This is appropriate in instances of lagophthalmos and persistence of corneal epithelial defects.
The chronic phase of ocular involvement in Stevens-Johnson syndrome necessitates caring for the consequences of the acute exanthem: punctal occlusion for sicca syndrome, electro- or cryoepilation for trichiasis, soft or scleral contact lens therapy for chronic keratopathy arising from residual trichiasis and from tarsal conjunctival keratinization, mucous membrane grafting, and corneal surgery for preservation of globe integrity and, in rare instances, for attempted visual rehabilitation.91
Recurrent SJS may respond to chronic systemic acyclovir therapy if herpes simplex virus is the agent provoking the attacks. Some cases of recurrent SJS not associated with herpes have responded to long-term systemic immunosuppressive chemotherapy.79
TOXIC EPIDERMAL NECROLYSIS
Definition
Toxic epidermal necrolysis (TEN) (Lyell’s disease, Ritter’s disease, scalded skin syndrome) is an acute systemic illness associated with a bullous eruption of the skin and mucous membranes that is, in many respects, similar to Stevens-Johnson syndrome.92,93,94 Lyell’s disease presents with a prodromal fever accompanied by malaise and tender or painful skin. The skin of the face, trunk, and extremities becomes inflamed; within 6 to 24 hours, the epidermis separates in large sheets (Fig. 16). Large, flaccid bullae may develop within the epidermis; these are mobile, and Nikolsky’s sign is positive. In contrast, the bullae that are present in Stevens-Johnson syndrome are subepidermal in location and are tense and nonmobile.94 The bullae of Lyell’s disease usually remain sterile, even in children in whom staphylococcal infection is associated with the disease. Therefore, it is not a pyoderma. The mortality rate is approximately 10%, and most of these fatalities are in infants.
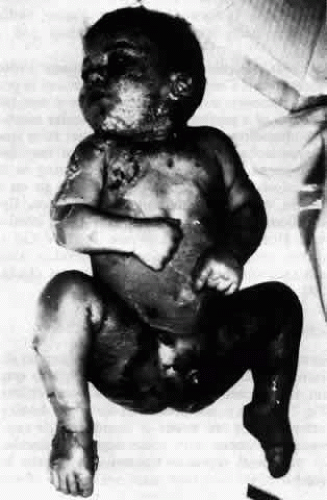 Fig. 16. Toxic epidermal necrolysis. (Courtesy of Dermatology Service, WRAMC) |
Epidemiology
TEN has no ethnic or geographic predilections, and it can affect patients of any age. The adult type of disease is more commonly associated with drug ingestion and affects all ages, although females are affected one and a half times more frequently than males.
Pathogenesis
The disease may affect all age groups, but when seen in infants and children under 10 years of age, it is usually associated with staphylococcal infection and is called Ritter’s disease (see Fig. 16). The most commonly involved organisms are phage group 2 staphylococci.92 The drugs often associated with this disease are penicillin, phenytoin, sulfonamides, phenolphthalein, and phenylbutazone. A case has been described in a patient on allopurinol therapy.95 The mortality rate in this group may be as high as 40%.93
Diagnosis
TEN secondary to drug or viral exposure is diagnosed by the history of such exposure or prodrome and the clinical picture of generalized bullae formation and exfoliation. Nikolsky’s sign is positive, and lesion biopsy discloses blister formation with a cleavage plane high in the epidermis, in the granular cell layer, with little associated inflammatory reaction.96
Ocular Manifestations
Ocular involvement in TEN is limited to the conjunctiva; it is usually less severe than is the ocular involvement in Stevens-Johnson syndrome. A mucopurulent conjunctivitis is the most common early manifestation, and, unlike the involvement in erythema multiforme, this does not usually result in ulceration, scarring, symblepharon, or corneal pannus formation.70 It may do so, however, with corneal erosions and all the other consequences described for SJS.93,94
Therapy
The acute and chronic therapy for the ocular involvement in TEN is identical to that described above for SJS.
EPIDERMOLYSIS BULLOSA
Definition
The term epidermolysis bullosa (EB) refers to a group of hereditary, and one acquired, bullous eruptions of the skin and mucous membranes that are characterized by the formation of blisters and erosions at sites of injury.97 There are several recognized forms of hereditary epidermolysis bullosa (Table 3). Ocular involvement may occur in many of these disorders, including the acquired variety, epidermolysis bullosa acquisita.98,99,100,101,102,103,104
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


