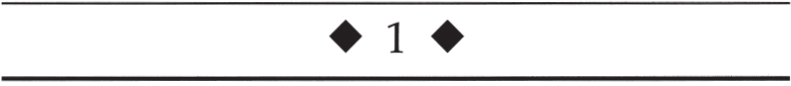
The Diagnosis of Optic Neuropathies
The diagnosis of an optic neuropathy is usually made on clinical grounds alone. Several excellent references discuss in detail the anatomy of the optic nerve as well as examination techniques (Burde, 1992; Miller, 1998; Trobe, 2001). The clinical features of optic neuropathies are summarized in Table 1–1. Other more sophisticated (and time-consuming) tests for optic neuropathy, such as visual evoked potentials, flicker fusion, formal color vision testing, and contrast sensitivity, are not discussed.
Once the diagnosis of optic neuropathy has been made, it is important to consider a wide differential diagnosis of possible etiologies, including hereditary, inflammatory, infiltrative, ischemic, demyelinating (optic neuritis), toxic, and compressive optic neuropathies. We refer the reader to the specific chapter on each type of optic neuropathy for further details.
Can the Appearance of the Optic Nerve Differentiate Etiology?
In general, the appearance of the optic nerve (e.g., normal, swollen, or pale) is not specific and cannot differentiate among various possible etiologies for optic neuropathy. Trobe et al reviewed 163 color fundus photographs of several entities resulting in optic atrophy, including glaucoma, central retinal artery occlusion (CRAO), ischemic optic neuropathy (ION), optic neuritis (ON), hereditary optic neuropathy (Leber’s and non-Leber’s types), compressive optic neuropathy (CON), and traumatic optic neuropathy (TON) (Trobe, 1980). These photographs were reviewed by five ophthalmologists as “unknowns.” Glaucoma, CRAO, and ION were correctly identified as the etiology by at least one of the five observers with an accuracy above 80%, but the remaining etiologies were correctly identified in less than 50% of cases! Helpful features in differentiating the entities included:
1. The presence of retinal arteriolar attenuation and sheathing in ischemic lesions (e.g., CRAO or ION).
2. Temporal pallor in entities selectively involving central vision and central visual field with sparing of peripheral visual field (e.g., optic neuritis and toxic optic neuropathies).
3. Superior or inferior (sector) optic disc pallor in ION.
| Decreased visual acuity Decreased color vision Visual field defect Ipsilateral relative afferent pupillary defect in unilateral or bilateral, asymmetric cases Light-near dissociation of the pupils in bilateral and symmetric cases Optic disc edema or disc atrophy (although the optic nerve may appear normal in retrobulbar optic neuropathy) |
Although optic disc cupping was often identified in glaucoma, it was also seen in 20% of cases not associated with glaucoma. Optic disc cupping in glaucoma cases, however, was more profound than in nonglaucomatous cases and greater neuroretinal rim pallor occurred in the nonglaucomatous cases. In patients with glaucoma, there is often absence of at least part of the neuroretinal rim/and the color of the remaining rim is normal. With nonglaucomatous optic neuropathy, rarely is any area of the rim completely absent and the remaining rim is often pale. Interestingly, only 11% of these cases with a known history of papillitis or ION had sufficient clues to identify previous disc swelling (Trobe, 1980).
Another study suggested that optic disc appearance may help differentiate anterior ischemic optic neuropathy (AION) from ON, although there are overlapping features. Optic disc stereographs were reviewed by masked observers (87 AION and 68 ON) (Warner, 1997). Altitudinal disc swelling was more than three times more common in AION than ON, although most discs were diffusely swollen. Most patients with AION had hemorrhages, whereas most ON cases did not. Almost all discs with ON had normal color or were hyperemic; only 35% of discs with AION had pallid swelling. Pallid swelling was so rare in ON, however, that of discs with pallor, 93% had AION. Arterial attenuation was also much more typical of AION. AION was the clinical diagnosis in 82% of cases with altitudinal edema, 81% of cases with disc hemorrhage, 93% of cases with pallid edema, and 90% of cases with arterial attenuation. A pale optic nerve with hemorrhage, regardless of type of edema, always represented AION (100%). A normal color nerve without hemorrhage reflected ON in 91% of cases, increased from only 76% if hemorrhage was not considered. A hyperemic nerve with hemorrhage represented AION in 82% of cases, but if altitudinal edema was also present, AION incidence increased to 93%.
In addition, numerous authors have stressed the localizing value to the optic chiasm or optic tract of a special type of optic atrophy caused by specific involvement of the nerve fiber layer of the nasal and temporal retina, respectively. Involvement of these fibers results in atrophy of the nasal and temporal optic disc with sparing of the inferior and superior poles (“band” or “bow tie” atrophy). Band atrophy occurs in the eye contralateral to the involved optic tract and may be unilateral or bilateral with lesions of the optic chiasm.
Neither the pattern (e.g., central scotoma, arcuate, altitudinal) of ipsilateral visual field impairment nor the severity of visual loss is pathognomonic for a specific optic neuropathy, and virtually any visual field defect may occur with any optic neuropathy (Trobe, 1978). In their report on 35 eyes in 20 patients with CON and 70 eyes in 54 patients with ON, Trobe and Glaser found central scotomas in 33% of cases of CON (vs. 75% in ON) and felt that a central scotoma could not be used as a differentiating feature between the two entities (Trobe, 1978).
The following sections describe the evaluation of optic neuropathy; this approach is summarized in Figure 1–1. We begin with an age-based differential diagnosis of an acute optic neuropathy. Two of the most common causes of acute optic neuropathy are AION and ON. Although there is considerable overlap in their clinical presentation, age can be used as an initial differentiating feature in many cases (Rizzo, 1991). In younger patients (< 40 years old) with acute unilateral optic disc edema and evidence for an optic neuropathy, ON is more likely than AION. Conversely, in the older patient with acute optic disc edema and visual loss, AION is more common (class III).
Is the Clinical Presentation Typical for Anterior Ischemic Optic Neuropathy?
The features of typical AION are discussed in Chapter 4. If these features are present, the patient should undergo an evaluation for underlying vasculopathic risk factors and giant cell arteritis (class III–IV, level B).
Is the Clinical Presentation Typical for Optic Neuritis?
The features and evaluation of typical ON are described in Chapter 2.
Is the Clinical Presentation Consistent with Optic Disc Edema with a Macular Star (ODEMS)?
The evaluation of optic disc edema with a macular star (ODEMS) is outlined in Chapter 3.
Is a Compressive Optic Neuropathy Present?
Compressive optic neuropathy (CON) usually causes painless, progressive, gradual loss of visual function (visual acuity, visual field, and color vision), a relative afferent pupillary defect (in unilateral or asymmetric cases), and optic disc edema or atrophy (but the optic disc may initially appear normal) (Bürde, 1992; Miller, 1998; Trobe, 1978). Unfortunately, CON may also present acutely or be steroid responsive and may masquerade as an inflammatory or demyelinating optic neuropathy.
CON that is due to orbital or intracanalicular lesions may result in ipsilateral optic disc edema followed by optic atrophy and may be associated with the development of abnormal blood vessels on the disc head called optociliary shunt vessels. These vessels probably represent collateral circulation between the retinal and choroidal venous circulation that allows venous blood to bypass the compression at the level of the optic nerve. The presence of an unexplained relative afferent pupillary defect or unexplained optic atrophy should prompt appropriate neuroimaging studies (usually magnetic resonance imaging of the involved optic nerve) (Guy, 1990). Orbital signs such as proptosis, Chemosis, or conjunctival injection should direct the imaging studies to the orbit (class III–IV, level B). Table 1–2 lists some possible causes of CON. Tables 1–3, 1–4, 1–5, and 1–6, and Figures 1–2 and 1–3, review the main clinical features of meningioma affecting the anterior visual pathways, optic nerve glioma, and craniopharyngioma.
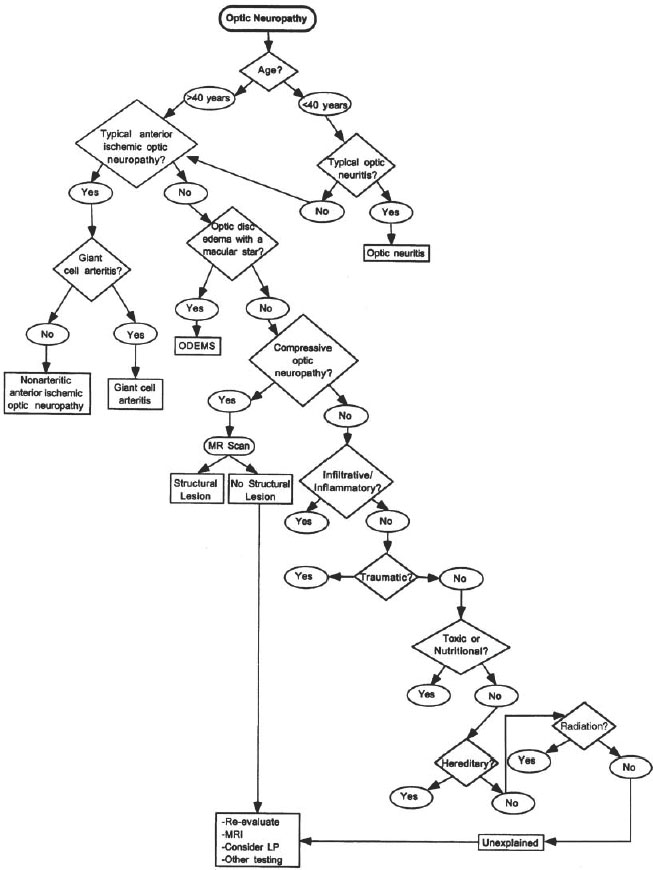
Figure 1–1. Evaluation of an optic neuropathy.
Is There Clinical Evidence for an Infiltrative or Inflammatory Optic Neuropathy?
Infiltrative or inflammatory optic neuropathy may present with the typical features of an optic neuropathy discussed above. As described in Chapter 2, the clinical profile of typical ON (e.g., pain with eye movement, typical age of onset, etc.) should be differentiated from atypical ON (e.g., lack of pain, atypical age of onset, anterior or posterior segment inflammation, etc.). Atypical cases should undergo an evaluation for infiltrative or inflammatory etiologies as listed in Table 1–7 (class IV, level C).
Intracranial or intraorbital benign and malignant tumors (Burde, 1992; Golnik, 1996; Katz, 1991; Kazim, 1992; Kodsi, 1993; Lee, 1997b) Meningioma (see Table 1–3) Glioma (see Tables 1–4 and 1–5) Craniopharyngioma (see Table 1–6) Pituitary adenoma Lymphoma and leukemia (Brazis, 1995; Nygaard, 1991; Park, 1993; Roth, 2000) Germinoma (Nakajima, 2001) Sinus histocytosis with lymphadenopathy (Goldberg, 1998) Nasopharyngeal cancer Metastasis (Kattah, 1993; Newsom, 1999; Pengel, 1997) Extramedullary hematopoiesis (Aarabi, 1998) Orbital fractures Pneumatocele (Wein, 1999) Inflammatory or infectious diseases (e.g., mucoceles, sclerosing orbital inflammation) (Hao, 1994; Loehrl, 2000; Thorne, 2002; Yamaguchi, 1997) Idiopathic hypertrophic cranial pachymeningitis (Tamai, 2000) Primary bone diseases (e.g., osteopetrosis, fibrous dysplasia, craniometaphyseal dysplasia, fibrosclerosis, Paget’s disease, aneurysmal bone cyst, pneumosinus dilatans, etc.) (Arroyo, 1991; Bland, 1992; Bocca, 1998; Caldermeyer, 1995; Chen, 1997; Daly, 1994; Grimm, 1995; Joseph, 1995; Katz, 1998; Michael, 2000; Saito, 1990; Schaffler, 2000; Skolnick, 2000; Steel, 1995; Stretch, 1992; Weisman, 1990) Vascular etiologies Orbital hemorrhage (Amrith, 1990; Buus, 1990; Dolman, 1991; Moorthy, 1992; Muthukumar, 1997) Orbital venous anomalies Carotid artery and anterior communicating artery aneurysms (Bakker, 1999; Miller, 1995; Misra, 1991; Ortiz, 1991; Shutter, 1993; Vargas, 1994) Dolichoectasia of the carotid artery (Colapinto, 1996; Jacobson, 1999; Savy, 1996) Compression by supraclinoid carotid artery (Ishikawa, 2000; Jacobson, 1999) Arteriovenous malformations Thyroid ophthalmopathy (see Chapter 16) Hydrocephalus Iatrogenic Intracranial catheters (Shults, 1993) Intranasal balloon catheter Intracranial oxidized cellulose hemostat Postoperative (e.g., post-optic canal decompression, sinus surgery) (Carter, 1998; Edelstein, 1998) Muslinoma (Bhatti, 2000; Lee, 1997a) |
Patients with inflammatory autoimmune optic neuropathy often have a progressive or recurrent steroid responsive or steroid dependent clinical course. A more detailed discussion of the evaluation of atypical ON and these alternative etiologies is found in Chapter 2. In patients with a possible inflammatory or infiltrative optic neuropathy, a lumbar puncture and additional laboratory studies (e.g., complete blood count, syphilis serology, antinuclear antibody, Lyme titer, chest radiograph, etc.) should be considered. The appropriate specific laboratory studies should be directed by pertinent history and examination findings. Table 1–8 reviews the evaluation of an atypical or unexplained optic neuropathy (class IV, level C).
Most commonly middle aged (peak in the 5th decade) Female: male = 3:1 White > African-American Increased frequency in neurofibromatosis May grow in pregnancy Symptoms Painless (rarely retro-orbital pain) Gradually progressive loss of vision or visual field defects If frontal, may have mental status changes May have diplopia if cavernous sinus involvement Olfactory groove may have anosmia
Ophthalmic signs May have relative afferent pupillary defect (RAPD) Optic disc edema (including papilledema) and/or optic atrophy May see optociliary shunt vessel (disc collaterals, visual loss, and optic atrophy—characteristic triad) Indocyanine green videoangiography may show abnormal hemodynamics of choroidal circulation in patients with sheath meningiomas (Muci-Mendoza, 1999) Visual acuity loss or visual field defects Generalized depression or constriction (orbital/canal/sphenoid) Central, paracentral, or cecocentral (orbital/canal) Homonymous hemianopsia (suprasellar/sphenoid) Bitemporal hemianopsia (suprasellar/sphenoid) May have proptosis (orbital/sphenoid) Motility deficits Sixth nerve palsy (most common), but any ocular motor palsy (third, fourth, sixth, combination) Restrictive extraocular muscle mechanical limitation if orbital lesion Paretic pattern if suprasellar/sphenoid/cavernous sinus Differential diagnosis of optic nerve sheath meningioma Sarcoidosis and other granulomatous diseases Optic nerve sheath meningocele (Garrity, 1990) Idiopathic hypertrophic cranial pachymeningitis Idiopathic inflammatory perioptic neuritis Metastasis (e.g., breast cancer) and other tumors (Newman, 1996) |
Source: Al-Mefty, 1990; Cunliffe, 1992; DeMonte, 1994; Dutton, 1991, 1992; Fayaz, 1999; Fineman, 1999; Garrity, 1990; Goldsmith, 1994a,b; Grunberg, 1991; Hirsch, 1993; Kinjo, 1995; Klink, 2000; Kotapka, 1994; Larson, 1995; Lee, 1996; Lee Wan, 1990; Lundsford, 1994; Mafee, 1999; Maroon, 1994; Moyer, 2000; Muci-Mendoza, 1999; Newman, 1994, 1996; Rubinstein, 1994; Sadun, 1993; Stafford, 1998; Vaphiades, 2001; Weaver, 1993; Wilson, 1994; Wroe, 1991; Zimmerman, 1990b.
Age Can present at any age Usually < 10 years old (75%) Mean 8.8 years (90% < 20 years old) No gender predilection Association with neurofibromatosis type 1 (NF1) 29% of optic nerve gliomas occur in setting of NF1 15% of NF1 who have no visual symptoms have glioma Patients with NF1 may have borderline favorable or no different prognosis than patients without NF1 Location of infiltration (topographic localization) One or both optic nerves (nerve alone in 24%) Optic disc (1.6%) Optic chiasm (75.7%) or tract In general, the more anterior the lesion, the better the prognosis Signs and symptoms Proptosis Painless progressive visual loss (optic neuropathy) Visual loss at presentation in 87.5% Hypothalamic symptoms (26%) or endocrinologic: diabetes insipidus, diencephalic wasting, precocious puberty, somnolence, growth failure Disc swelling (35%) or atrophy (59%) Rare optociliary shunt vessels Strabismus Nystagmus (23%) (spasmus nutans-like nystagmus) Visual field defects (central or bitemporal) Headache (23%) Intrinsic enlargement of optic nerve with variable contrast enhancement Neuroimaging Magnetic resonance (MR) scan with gadolinium superior to computed tomography (CT) scan Differential diagnosis Parenchymal optic nerve enlargement Sarcoid Tuberculosis Syphilis Optic neuritis (e.g., multiple sclerosis) Optic nerve infiltration (e.g., leukemia, lymphoma) Extraparenchymal Optic nerve sheath meningioma Optic nerve sheath metastasis Treatment Controversial Gliomas are often static lesions May enlarge and cause progressive visual loss in one or both eyes Treatment recommendations outlined in Figure 1–3 (Lee, 1999) Observation in stable cases is reasonable (class III–IV, level B) Radiation therapy (class III–IV, level C) Usually 5400 cGy in daily dose fractions 150-180 cGy Radiation risks include cerebrovascular disease, moyamoya disease, cerebral atrophy, subnormal intelligence or learning disabilities, secondary malignancies (e.g., astrocytomas), cataracts, radiation retinopathy or optic neuropathy, endocrinopathy, hypothalamic dysfunction Chemotherapy—various agents in various combinations: actinomycin-D, vincristine, 2 chloroethyl-cyclohexyl-1-nitrosourea (CCNU), 6-thioguanine, procarbazine, dibromodulatol, topotecan, carboplatin, etoposide (class III–IV, level C) Surgical therapy (class III–IV, level C) Optic nerve glioma with no useful vision or progression may be resected Chiasmal hypothalamic, optic tract glioma cannot be resected Exophytic component of tumor may be debulked Hydrocephalus may require shunting procedure Prognosis 80% have stable vision after an initial period of visual loss 10-year survival rate 85–100% in various series Spontaneous regression may occur |
Source: Brodovsky, 1997; Chateil, 2001; Créange, 1999; Cummings, 2000; Deliganis, 1996; DiMario, 1993; Drake, 1991; Dunn, 1990; Dutton, 1994; Epstein, 1992; Friedman, 1997; Fuss, 1999; Garvey, 1996; Gayre, 2001; Grill, 1999; Hoffman, 1993; Imes, 1991; Janss, 1995; Jenkin, 1993; Kestle, 1993; Kovalic, 1990; Levin, 1992; Listernick, 1992, 1994, 1997; Liu, 1992a, 2001; Moghrabi, 1993; Nishio, 1993; Oaks, 1990; Packer, 1993, 1994; Parsa, 2001; Petronio, 1991; Pierce, 1990; Rodriguez, 1990; Shuper, 1997; Sutton, 1994, 1995; Wisoff, 1990a,b.
Age at presentation: middle-age; range 6–79, mean 47.8; 73% were 40 or older Sex: 65% males and 35% females Clinical signs and symptoms Decreased vision Bilateral or unilateral Visual acuity usually falls to blindness over average of 11.1 weeks (range 1–60 weeks) Optic nerve visual held defects Normal discs, optic disc swelling or atrophy Proptosis Ophthalmoplegia Retro-orbital pain common Macular edema, cherry-red spot, and flame hemorrhage or hemorrhagic papillopathy may simulate central retinal vein occlusion (CRVO) Not associated with NF1 (neurofibromatosis) Location Involves chiasm and at least one contiguous optic nerve; often involves hypothalamus, third ventricle, basal ganglia, temporal lobe Primarily affects chiasm and intracranial optic nerves Treatment Radiation Chemotherapy Treatment may temporarily improve or rarely stabilize vision Pathology: malignant astrocytoma Prognosis Poor Overall mortality 97% Mean survival 8 7 months (3 to 24 months) |
Is There Evidence for Traumatic Optic Neuropathy?
The features and evaluation of TON are discussed in Chapter 6.
Is There Evidence for a Toxic or Nutritional Optic Neuropathy?
Patients with toxic optic neuropathies usually present with painless, bilaterally symmetric, and slowly progressive visual loss The visual field defect is typically bilateral central or cecocentral scotomas The optic nerves may appear normal until late in the course of the disease when optic atrophy (often temporal pallor) usually develops Occasionally the discs may be swollen and slightly hyperemic A number of medications and toxins may result in optic neuropathy (Brazis, 1998; Danesh-Meyer, 2000; Sedwick, 1991,1992) These are summarized in Table 1–9 Most of these etiologies can be excluded by a careful and detailed exposure and occupational history.
Any age Bimodal incidence Peak age < 20 and 50 to 70 years old Equal sex distribution Clinical Decreased visual acuity (optic nerve, chiasm, optic tract) In children—often decreased acuity and papilledema (50%) In adults—less commonly papilledema Signs of increased intracranial pressure (headache, nausea, vomiting) Endocrine Absent or precocious sexual development Growth disturbances Variable hypopituitarism Diabetes insipidus Obesity Impotence Amenorrhea/galactorrhea Somnolence, confusion, or dementia (especially in older patients) Ocular findings Seesaw nystagmus Visual held defects Inferior bitemporal field defect (most have field defects) May have incongruous, asymmetric defect May involve optic tract May cause ocular motor nerve palsies Neuroimaging Magnetic resonance imaging (MRI) delineates tumor and intracranial anatomy Computed tomography (CT) shows calcification better Occasionally may infiltrate optic nerve, chiasm tract, mimicking primary intrinsic tumor such as optic glioma (“potbelly” appearance of optic nerve) Treatment Surgical: complete vs. partial resection Radiotherapy Cyst aspiration and P32 instillation Consider intracystic chemotherapy (bleomycin) Secondary malignant glioma can develop after radiation therapy |
Source: Brummitt, 1992; Crotty, 1995; El-Mahdy, 1998; Fahlbusch, 1999; Honegger, 1999; Petito, 1996; Rao, 1995; Weiner, 1994; Youl, 1990
Ethambutol is a commonly used medication that may cause toxic optic neuropathy The mechanism of ethambutol toxicity is poorly understood but may be related to zinc depletion (Schild, 1991) The incidence of toxicity is dose and duration dependent (Choi, 1997; Harcombe, 1991; Kumar, 1993; Russo, 1994; Schild, 1991; Seth, 1991; Thomas, 1994; Tsai, 1997), with the incidence of optic neuropathy being as high as 6% at doses of 25 mg/kg/day Doses less than 15 mg/kg/day are thought to be relatively safe, but optic neuropathy may occur even at “safe” doses.
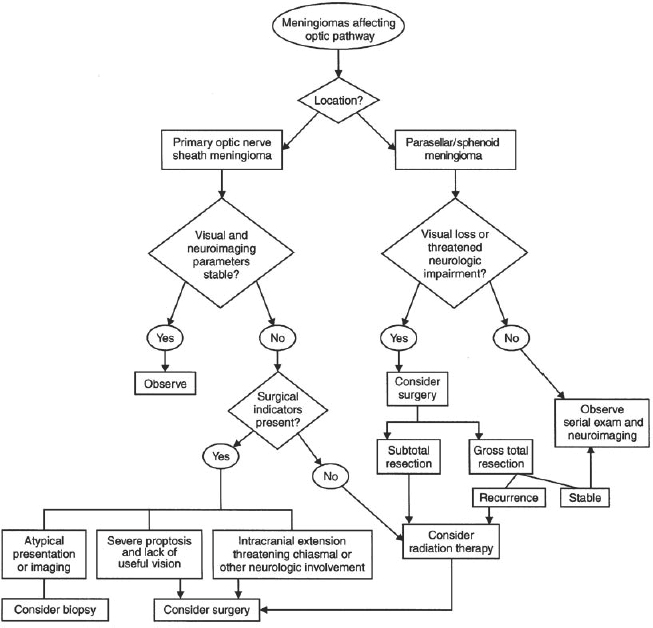
Figure 1–2. Treatment algorithm for meningiomas affecting optic pathway.
Barron et al reported ethambutol optic neuropathy in 3 of 304 (0.99%) patients treated with ethambutol at 25 mg/kg/day for 60 days followed by 15 mg/kg/day (Barron, 1974). Leibold described two types of visual loss due to ethambutol toxicity: a central toxicity (e.g., decreased visual acuity, central scotomas, and impaired color perception) and a periaxial toxicity (e.g., normal or almost normal visual acuity, normal color perception, and peripheral quadrantic scotomas or constriction) (Leibold, 1966). There was a 20% incidence of central toxicity and an 11% incidence of periaxial toxicity in 35 patients receiving doses higher than 35 mg/kg/day for a minimum of 185 days. A 5.3% incidence of periaxial toxicity occurred in the 38 patients receiving less than 35 mg/kg/day (Leibold, 1966). Although many authors feel that doses of 25 mg/kg/day /day for less than 2 months followed by maintenance doses of 15 mg/kg/day are safe, there are cases of visual loss even at “safe” doses (Alvarez, 1993; Thomas, 1994; Tsai, 1997). Bronte-Stewart et al reported five patients with severe visual loss after 25 mg/kg/day for 2 months followed by 15 mg/kg/day (Bronte-Stewart, 1976). Three of these five patients had renal disease that may have increased drug levels because 70% of the ethambutol dose is excreted by the kidneys (Citron, 1986). Tsai and Lee reported 10 patients with ethambutol optic neuropathy from “safe” doses, stressing that there is in fact no safe dose of ethambutol. Toxicity in this study was most prominent in individuals over the age of 60 years, and thus this drug must be used with caution, especially in elderly patients (Tsai, 1997). Isoniazid (isonicotinic acid hydrazide, INH), especially in combination with ethambutol, has also been reported to cause a toxic optic neuropathy, and isoniazid toxicity should be suspected as the etiology in cases of persistent visual loss despite discontinuation of ethambutol (Jimenez-Lucho, 1987). Visual evoked potential studies may be useful in evaluating patients with early ethambutol toxicity (Kumar, 1993).
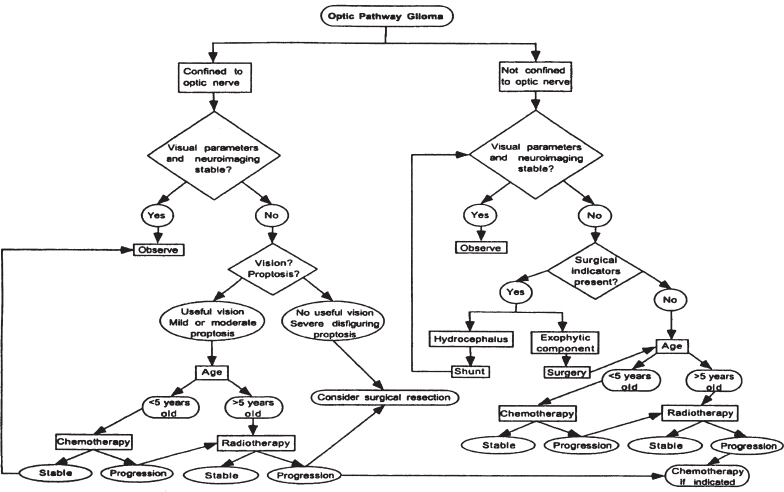
Figure 1–3. Treatment algorithm for optic pathway gliomas based on location. (Reprinted from Lee, 1999, with permission from © Swets & Zeitlinger.)
Nutritional deficiencies may result in optic neuropathy (Bourne, 1998; Lessell, 1998). Some vitamin and nutrient deficiencies causing an optic neuropathy are listed in Table 1–10.
Pernicious anemia or dietary deficiency (e.g., vegetarian) may result in a vitamin B12 deficiency optic neuropathy. The pathophysiology of “alcohol amblyopia” is probably related to a deficiency (nutritional amblyopia) of B12, thiamine, and/or folate (rather than a direct toxic effect of alcohol). The ability of tobacco alone to cause a toxic optic neuropathy has been asserted by several authors (Samples and Younge, 1981). Samples and Younge (1981), for example, state that central and cecocentral scotomas may occur in association with smoking alone, especially cigar smoking. A toxic effect of cyanide may be the basis for tobacco optic neuropathy (Bronte-Stewart, 1976). Smoking may also impair intestinal vitamin B12 absorption.
Patients suspected of harboring a toxic or nutritional optic neuropathy should be screened for nutritional deficiencies and treated with appropriate supplementation (class IV, level C). These patients should be urged to discontinue alcohol and tobacco use. Both serum and erythrocyte folate levels should be checked because there may be variability in the serum folate level alone (especially related to recent meals) (Golnik, 1994).
Neoplastic Plasmacytoma and multiple myeloma (Maini, 1997) Carcinomatous meningitis (Freilich, 1995; Ing, 1996; Katz, 1991; McFadzean, 1994; Sung, 1998; Teare, 1991) Leukemia (Brown, 1992a; Camera, 1993; Costagliola, 1992; Cramer, 1996; Horton, 1992; Pierro, 1992; Shibasaki, 1992; Wallace, 1991) Lymphoma (Dunker, 1996; Fierz, 2001; Forman, 1998; Guyer, 1990; Noda, 1993; Siatkowski, 1992; Strominger, 1993; Yamamoto, 1994; Zaman, 1993) Infiltrative orbitopathy in POEMS syndrome Reactive lymphocytosis with pseudolymphoma from phenytoin (Galetta, 1991) Paraneoplastic disease (Ing, 1996; Lieberman, 1999; Luiz, 1998; Malik, 1992; Oohira, 1991; Thambisetty, 2001) Idiopathic hypertrophic cranial pachymeningitis (Aylward, 1995; Botella, 1994; Girkin, 1998; Hamilton, 1993; Jacobson, 1996; Kawano, 1995; Lam, 1994; Levine, 1993; Mamelak, 1993; Nishizaki, 1997; Olmos, 1993; Parney, 1997; Rootman, 1994) Infectious etiologies Cryptococcal meningitis (Cohen, 1993) Aspergillus (Brown, 1994; Dinowitz, 2001; Hutnik, 1997; Johnson, 1999) Mucormycosis (Balch, 1997) Cysticercosis (Chandra, 2000; Gulliani, 2001; Gurha, 1999) Lyme disease (Lesser, 1990) Tuberculosis Toxoplasmosis (Song, 2002) Syphilis (Danesh-Meyer, 1999) Cat-scratch disease (Golnik, 1994b) HIV (AIDS) (Cacciatori, 1996) Inflammatory diseases (Burde, 1992) Churg-Strauss (Acheson, 1993) Contiguous sinus disease Behçet’s disease Sarcoidosis (Achiron, 1995; Beck, 1994; Carmody, 1994; DeBroff, 1993; Ing, 1997; Kosmorsky, 1996; Pelton, 1999; Sharma, 1991; Silver, 1994; Thorne, 1998) Wegener’s granulomatosis (Beiden, 1993) Systemic lupus erythematosus (Ahmadieh, 1994; Rosenbaum, 1997; Siatkowski, 2001) Sjögren’s syndrome Relapsing polychondritis Polyarteritis nodosa Inflammatory bowel disease Granulomatous hypophysitis (Arsava, 2001) Isolated optic nerve pseudotumor (Patankar, 2000) Sclerosing orbital inflammation (Thorne, 2002) |
Toxic or nutritional optic neuropathies are painless, subacute in onset, and bilateral, and usually involve central visual acuity and visual fields (e.g., central and cecocentral scotomas), but their clinical presentations may be variable. Unfortunately, CON may mimic the clinical presentation of toxic optic neuropathy, and neuroimaging is recommended. The determination of presumed toxic or nutritional optic neuropathy should include a complete evaluation to exclude other etiologies of bilateral, painless, and progressive optic neuropathies (e.g., hereditary optic neuropathy, bilateral compressive optic neuropathy, etc.). The evaluation of presumed toxic optic neuropathy is outlined in Table 1–11 (class IV, level C).
First-line testing Magnetic resonance imaging of optic nerve(s) Erythrocyte sedimentation rate Complete blood count with differential Syphilis serology Antinuclear antibody (ANA) Chest radiograph Angiotensin-Converting enzyme (antineutrophil cytoplasmic antibody, ANCA) Lumbar puncture Second-line testing Gallium scan if sarcoidosis suspected Purified protein derivative (PPD) skin testing if tuberculosis suspected Anti-double-stranded DNA, complement levels, etc., if systemic lupus erythematosus or other collagen vascular disease suspected Leber’s hereditary optic neuropathy mutation blood test Heavy metal screen Serum vitamin B12 and folate levels Lyme titer if endemic area or exposure history Paraneoplastic antibody profile (e.g., autoantibodies for collapsin response mediated protein (CRMP)-5 may be associated with optic neuropathy in patients with lung cancer, especially small-cell type, or thymoma) (Cross, 2002; Thambisetty, 2001; Yu, 2001) Consider more specific serologic studies if infectious process suspect (e.g., Bartonella titers for cat-scratch disease, toxoplasmosis titers, toxocara titers, etc.) |
Is There a History of Radiation Exposure to the Optic Nerves?
Radiation optic neuropathy (RON) is thought to be an ischemic disorder of the optic nerve that usually results in irreversible severe visual loss months to years after radiation therapy to the brain or orbit (Arnold, 1995; Borruat, 1993, 1996; Ebner, 1995; Girkin, 1997; Glantz, 1994; Goldsmith, 1992; Guy, 1991, 1995; Hudgins, 1992; Jiang, 1994; Landau, 1996; Leber, 1998; Liu, 1992; McClellan, 1995; Parsons, 1994; Polak, 1995; Roden, 1990; Tachibana, 1990; Young, 1992; Zimmerman, 1990). It is most often a retrobulbar optic neuropathy, and thus the optic nerve may appear normal on initial examination. Approximately three fourths of patients have bilateral involvement. The visual loss is characteristically rapid and progressive, with the disc becoming pale over a period of 4 to 6 weeks. Final vision is NLP in 45% and worse than 20/200 in an additional 40% of affected eyes (i.e., 85% of eyes with RON have a final visual acuity of 20/200 or worse). More rarely, RON may present as an anterior optic neuropathy with optic disc swelling (Parsons, 1994). Such cases usually occur in the setting of radiation retinopathy following treatment of orbital or intraocular lesions. Associated findings of radiation retinopathy resemble those of diabetic retinopathy and variably include peripapillary hard exudates, hemorrhages, subretinal fluid, cotton-wool spots, focal arteriolar narrowing, macular edema, capillary nonperfusion, capillary telangiectasia, microaneurysms, neovascularization of disc and retina, perivascular sheathing, vitreous hemorrhage, neovascular glaucoma, central retinal artery occlusion, and central retinal vein occlusion. Loss of vision with anterior cases may be due to macular edema, macular hemorrhages, macular exudates, or perifoveal capillary nonperfusion, as well as from optic nerve involvement. The clinical features of RON are outlined in Table 1–12.
Common etiologies Ethambutol—tuberculosis therapy (Harcombe, 1991; Kumar, 1993; Russo, 1994; Schild, 1991; Seth, 1991; Thomas, 1994; Tsai, 1997) Ethanol and tobacco (tobacco alcohol amblyopia) (Danesh-Meyer, 2000; Sedwick, 1991, 1992) Less common etiologies Amantadine—antiviral, Parkinson’s disease Amiodarone (Cardarone)—cardiac disease (Macaluso, 1999; Sedwick, 1992; Speicher, 2000; Sreih, 1999) Amoproxan—vasodilator and antiarrhythmic Aniline dyes Aspidium (male fern) Barbiturates—sedative, anticonvulsant Cafergot—headache Carbon disulfide—manufacture of viscose rayon fibers and cellophane films Carbon monoxide (Simmons, 1998) Carbon tetrachloride—manufacturing of refrigerants and aerosols, dry-cleaning fluid, fat solvent, fire extinguishers, insecticides, shampoo Cephaloridine—antibiotic Chloramphenicol—antibiotic (Thomas, 1994) Chloronitrobenzene and dinitrobenzene—explosives Chlorpromazine (Thorazine)—antipsychotic Chlorpropamide (Diabenese)—diabetes Cimetidine (Sa’adah, 1999) Ciprofloxacin (Cipro)—antibiotic (Vrabec, 1990) Cisplatin plus carboplatin—chemotherapy (Caraaceni, 1997) Cisplatin plus carmustine—chemotherapy (Wang, 2000) Clioquinol—antibiotic Cobalt chloride Corticosteroids (Teus, 1991) Cyanide intoxication (dietary) Cyclosporine—chemotherapy (Avery, 1991) D-penicillamine—rheumatologic Deferoxamine—for removal of excess iron in patients requiring long-term transfusions (Pinna, 2001) Dichlorodiphenyltrichloroethane (DDT)—insecticide Digitalis (Digoxin)—cardiac disease Diiodohydroxyquin—amoebocide Dinitrotoluene—explosive Disulfiram (Antabuse)—alcohol addiction Elcatonin—synthetic analogue of calcitonin (Kimura, 1996) Emetine—amoebocide Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|