The Developmental Glaucomas
Terri-Diann Pickering
Patricia C. Wong
Christopher J. Dickens
H. Dunbar Hoskins Jr
The developmental glaucomas are a group of diseases characterized by maldevelopment of the eye’s aqueous outflow system. The resulting elevated intraocular pressure may occur at birth or anytime thereafter. Early recognition and therapy can significantly improve a child’s visual outcome.
This chapter uses the Shaffer-Weiss disease classification, which divides the developmental glaucomas into the more common primary congenital glaucoma and glaucoma associated with other ocular or systemic congenital anomalies (Table 1).1 The recognition of associated syndromes can be helpful in genetic counseling. A third category seen in infancy consists of glaucomas that are not truly congenital but are acquired, secondary forms of glaucoma. These will be mentioned briefly.
TABLE 1. Shaffer-Weiss Classification of Congenital Glaucoma Patients | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
An anatomic classification defines the actual developmental defect that is clinically evident at the time of examination (Table 2).2,3,4 The identification of anatomic defects can be useful in determining appropriate therapy and prognostic factors.
TABLE 2. Hoskins’ Anatomic Classification of the Developmental Glaucomas | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Normal variations in the infant eye as well as the secondary changes from elevated intraocular pressure are discussed first to distinguish them from anatomic defects associated with developmental glaucoma.
NORMAL INFANT EYE
At birth, the normal infant eye has a clear cornea approximately 10 mm in diameter. The intraocular pressure is usually below 16 mm Hg. The iris has a thin anterior stroma in the periphery that is not hypoplasia of the anterior stroma but incomplete development of the iris; this thickens as the child matures.
In the newborn, the iris inserts into the ciliary body, posterior to the scleral spur. The ciliary body is seen as a distinct band immediately anterior to the iris insertion. The presence of this band usually distinguishes the normal infant eye from one with congenital glaucoma associated with trabeculodysgenesis. Insertion of the infant iris into the angle wall is flat compared to the adult angle recess configuration. Recession of the angle, which turns the iris posteriorly before inserting into the ciliary body, does not occur until the first 6 to 12 months of life.
EFFECTS OF ELEVATED INTRAOCULAR PRESSURE ON THE INFANT EYE
During the first 3 years of childhood, the collagen fibers of the eye are more elastic than later in life. Elevation of the intraocular pressure causes rapid enlargement of the globe with progressive enlargement of the cornea and increase in axial length.5 As the cornea enlarges, stretching leads to ruptures of Descemet’s membrane, epithelial and stromal edema, and corneal clouding. The iris is stretched so that the stroma appears thinned. The scleral ring, through which the optic nerve passes, also enlarges with elevated intraocular pressure. This results in rapid cupping of the optic nerve, which can quickly reverse if the intraocular pressure is normalized. This reversal in cupping is not seen in adult eyes and is probably related to the greater elasticity of the optic nerve head connective tissues in the infant.6,7
Infant eyes with advanced disease demonstrate atrophic changes throughout all structures. The iris, trabecular meshwork, ciliary body, choroid, and retina are thinned and degenerated. The cornea is scarred because of chronic edema and ruptures of Descemet’s membrane. The optic nerve is completely cupped with little to no nerve tissue remaining.
ANATOMIC CLASSIFICATION OF DEVELOPMENTAL GLAUCOMAS
Maldevelopment of the anterior segment is the hallmark of developmental glaucoma. It may involve one or more of the angle structures: the trabecular meshwork, the iris, and/or the cornea.
ISOLATED TRABECULODYSGENESIS
In more than 50% of infants and juvenile patients presenting with glaucoma, isolated trabeculodysgenesis is the only developmental ocular anomaly seen. This anatomic defect is most commonly seen in primary congenital glaucoma.
There are no anomalies of the iris or cornea, although stretching effects secondary to elevated intraocular pressure may be seen. This trabecular maldevelopment presents in two forms. In the more common form, the iris inserts flatly into the trabecular meshwork at or anterior to the scleral spur (Fig. 1). This insertion usually obscures the ciliary body, although portions of the anterior ciliary body may be seen through a thickened trabecular meshwork if the angle is viewed obliquely. The level of iris insertion may vary along the angle circumference with portions of iris inserting behind the scleral spur and other portions inserting anterior to it. The surface of the trabecular meshwork may have a stippled, orange-peel appearance, and the peripheral iris stroma may appear thinned because of stretching from enlargement of the eye.
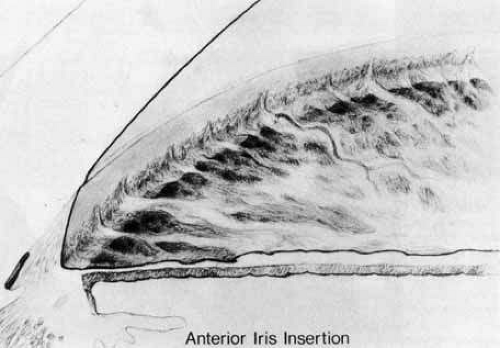 Fig. 1 Flat anterior iris insertion in trabeculodysgenesis. The iris inserts flatly and abruptly into the trabecular meshwork. Small iris processes are often visible on the trabecular surface. (Hoskins HD Jr., Shaffer RN, Hetherington J: Anatomical classification of the development of glaucomas. Arch Ophthalmol 102:1331, 1984. Copyright © 1984, American Medical Association.) |
The second form of isolated trabeculodysgenesis has a concave iris insertion. The plane of the iris is posterior to the scleral spur. However, the anterior stroma continues over the trabecular meshwork, obscuring the scleral spur and ending just posterior to Schwalbe’s line (Fig. 2).
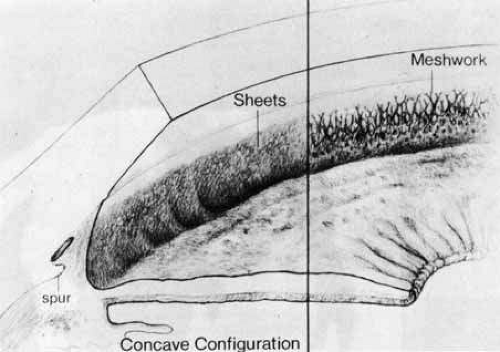 Fig. 2 Concave iris insertion in trabeculodysgenesis. Superficial iris tissue wraps around the angle recess and covers the internal surface of the trabeculum. This may take the form of dense sheets (left) or an arborizing network (right). This is different from the small processes seen in the anterior iris insertion. (Hoskins HD Jr, Shaffer RN, Hetherington J: Anatomical classification of the development of glaucomas. Arch Ophthalmol 102:1331, 1984. Copyright © 1984, American Medical Association.) |
IRIDOTRABECULODYSGENESIS
Iris anomalies may occur in addition to trabecular malformation. They may involve the anterior stroma, iris vessels, or full thickness of the iris. The trabecular meshwork appearance is similar to that found in isolated trabeculodysgenesis.
Anterior Stromal Defects
Hypoplasia of the anterior iris stroma is the most common iris defect associated with developmental glaucoma (Fig. 3). This disorder demonstrates malformation of the collarette with absence or marked reduction of the crypt layer. The pupillary sphincter may appear prominent. Hypoplasia of the anterior iris stroma may be seen in Axenfeld’s, Rieger’s, and Peters’ anomalies.
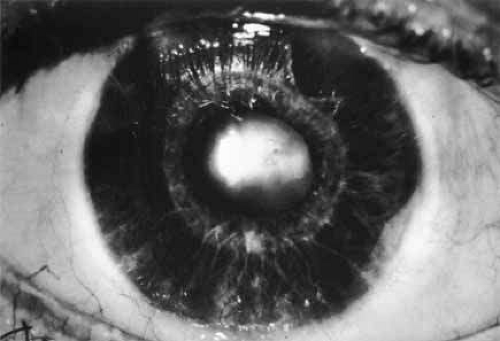 Fig. 3 Hypoplasia of the anterior iris stroma. The collarette is absent, and the sphincter is prominent. Note occasional strands of anterior stromal tissue above, and iris hole without sphincter involvement at the 9 o’clock position. (Surgical iridectomy at the 1 o’clock position.) |
In hyperplasia of the anterior iris stroma, there is a thickened, velvety, pebbled appearance. This is an uncommon condition and has only been seen in association with Sturge-Weber syndrome with glaucoma.
Anomalous Iris Vessels
Vascular anomalies of the iris can present as persistence of the tunica vasculosa lentis or as irregularly wandering superficial anomalous iris vessels. The first condition exhibits a regular arrangement of vessels looping into the pupillary axis in front of or behind the lens. Normal radial vessels on the iris surface are also prominent be-cause there is usually hypoplasia of the anterior iris stroma.
In the second condition, anomalous superficial iris vessels wander irregularly over the iris surface with an often distorted pupil (Fig. 4). The iris surface has a whorled appearance and the anterior iris stroma is often hypoplastic. These vascular anomalies differ from the normal exposure of radial iris vessels seen in lightly pigmented eyes. Anomalous iris vessels are seen most frequently in eyes that present with glaucoma and cloudy corneas at birth, and are not associated with any particular syndrome. It is unclear whether they represent an earlier onset of primary congenital glaucoma or an entirely different syndrome. They do indicate a more severe malformation of the anterior segment and a grave prognosis, with such cases requiring multiple surgeries.
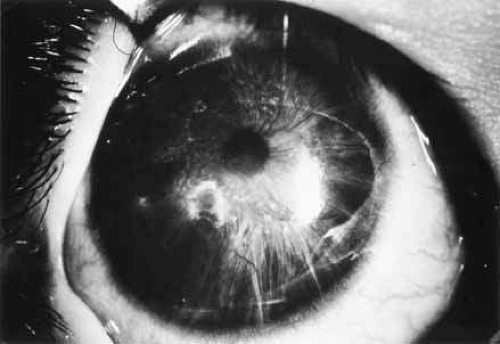 Fig. 4 Anomalous iris vessels. Vessels course irregularly over the iris surface. On the right, small vessels are seen crossing the pupillary margin. The pupil is distorted at the 10 o’clock position. Radial iris fibers are curved and irregularly arranged. |
Structural Iris Defects
These defects may present as full-thickness holes through the iris with or without sphincter involvement. A more extensive defect with the majority of iris absent is seen in aniridia (Fig. 5).
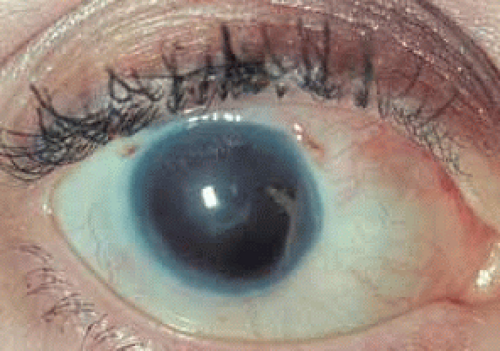 Fig. 5 Aniridia with cataract. The absence of iris tissue is made more obvious by the lens opacity. |
CORNEOTRABECULODYSGENESIS
Corneal stretching and clouding secondary to increased intraocular pressure are not considered congenital defects. Congenital corneal defects include peripheral, midperipheral, and central defects, as well as abnormalities in corneal size. There are associated iris abnormalities in the majority of cases.
Peripheral corneal lesions extend no more than 2 mm into clear cornea and usually involve the entire corneal circumference. The most common abnormality is Axenfeld’s anomaly, which is characterized by posterior embryotoxon and adherent iris tissue (Figs. 6 and 7).
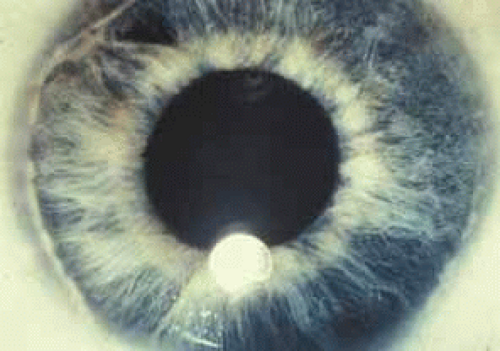 Fig. 6 Posterior embryotoxon. Note prominent Schwalbe’s line with iris attachments. (Courtesy of Robert N. Shaffer) |
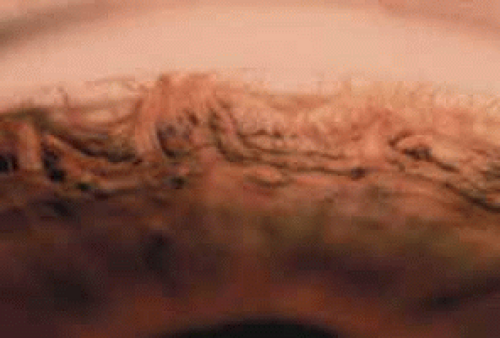 Fig. 7 Axenfeld’s anomaly. Gonioscopic view of iris adherent to Schwalbe’s line. (Courtesy of Robert N. Shaffer) |
Midperipheral corneal lesions involve broad areas of iris adhesion. The cornea is frequently opacified in these areas of attachment. Pupillary anomalies and iris holes are often associated. These lesions are generally found in Rieger’s anomaly.
Central corneal defects are usually opacified and thinned, with a zone of clear cornea between the defect and limbus. Strands of iris tissue from the collarette may attach to the leukoma. In some cases there may be lens apposition to the corneal defect. These findings are typically seen in Peters’ anomaly (Fig. 8).
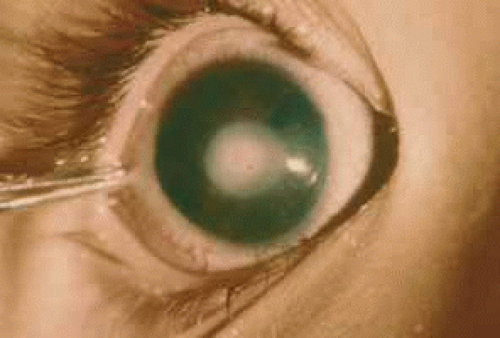 Fig. 8 Peters’ anomaly showing forward extension of the iris attached centrally to the opacified cornea. |
Abnormalities of corneal size present as microcornea or megalocornea. Microcornea may be seen in various congenital conditions, including microphthalmos, nanophthalmos, Rieger’s anomaly, and persistent hyperplastic primary vitreous. Megalocornea may be seen in patients with Axenfeld’s syndrome or as an X-linked recessive condition. Megalocornea must be distinguished from corneal enlargement caused by elevated intraocular pressure.
CLINICAL PRESENTATION
Signs and symptoms of glaucoma in an infant differ from those of an older child or adult because of the elastic response of the infant eye to elevated intraocular pressure. Infants often present with epiphora, photophobia, and blepharospasm because of corneal stretching, ruptures of Descemet’s membrane, and edema.10 The child may become irritable and so photophobic that he or she will bury his or her face to avoid light. Parents may notice haziness and enlargement of the eye and may initially consider the large eyes an attractive feature rather than a warning sign of glaucoma.
EXAMINATION
A thorough examination is usually best performed under general anesthesia. Delivery of anesthesia by mask with an oral airway is usually adequate for short examinations. However, if prolonged examination or surgery is required, endotracheal intubation should be performed.
After anesthesia is administered, the intraocular pressure should be checked as soon as is safely possible, because most general anesthetics lower intraocular pressure. The intraocular pressure may be measured with a hand-held electronic tonometer (Tono-pen), a Perkins hand-held applanation tonometer, a pneumotonometer, or a Schiötz tonometer.11,12 A useful upper limit is 21 mm Hg. However, this value is not absolute, and normal tolerable levels may vary among patients. Corneal changes caused by edema, calcification, or irregular corneal curvature can cause misleading intraocular pressure measurements.
The normal horizontal corneal diameter in a full-term newborn is 10 to 10.5 mm and increases to the adult diameter of approximately 11.5 to 12 mm by 2 years of age. A diameter greater than 12 mm in an infant is highly suggestive of congenital glaucoma.13 Measurements of the cornea are made in the horizontal meridian with calipers (Fig. 9). These recordings are accurate to 0.5 mm except when the limbus is stretched as a result of enlargement of the globe.
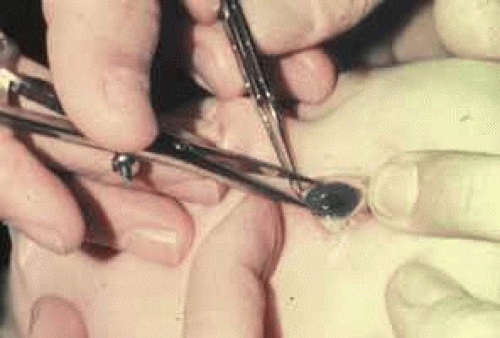 Fig. 9 Horizontal measurement of corneal diameter with calipers. |
Corneal clouding and breaks in Descemet’s membrane are also important in the evaluation of congenital glaucoma (Fig. 10). When corneal clouding is marked, it may be necessary to remove edematous corneal epithelium to view the other ocular structures. Developmental anomalies of the cornea and iris must be noted because they may alter diagnosis and treatment.
 Fig. 10 Enlarged cornea with breaks in Descemet’s membrane. |
Gonioscopy is performed by use of a gonioprism or a Koeppe lens with a Barkan light and binocular microscope. The Koeppe lens also aids in viewing the lens, vitreous, and fundus. It neutralizes irregular corneal surfaces and improves the view of the optic nerve through a small pupil, allowing the entire optic nerve head to be seen within one field.
Optic nerve evaluation is the most important part of the glaucoma evaluation. Glaucomatous disc changes occur more rapidly in infants and at lower pressures than in older children or adults. Cup-to-disc ratios greater than 0.3 are rare in normal infants and must be considered highly suspicious of glaucoma (Table 3).14
TABLE 3. Cup/Disc Ratios, Birth to Three Years | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
Asymmetry of optic nerve cupping is also suggestive of glaucoma, particularly differences greater than 0.2 between the two eyes. The glaucomatous cupping may be oval in configuration but is more often round and central. Enlargement of the cup is circumferential, with the nerve retaining its pink rim until advanced stages of cupping (Fig. 11).
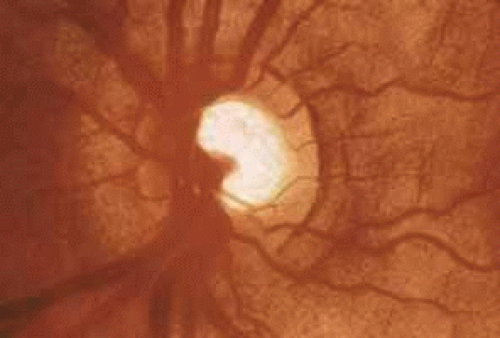 Fig. 11 Disc of glaucomatous infant with deep cupping. Pink tissue remains at the rim until the cupping is advanced. Note deep central cup with pink rim. |
The cup size will remain stable or decrease with successful intraocular pressure control. Such a reduction in cup size occurs most commonly in the first year of life. An increasing cup size indicates inadequate intraocular pressure control and progression of glaucoma. Therefore, it is very important for the clinician to make careful drawings or take photographs for future comparison. Some children may be followed by visual fields when they are older.15
Cycloplegic refraction and treatment of amblyopia should also be performed to allow the child the best possible visual outcome. There may be significant differences in refractive error, especially in cases of unilateral glaucoma. Greater myopia or less hyperopia is generally found in the involved eye.
PRIMARY CONGENITAL GLAUCOMA
Primary congenital glaucoma is the most common form of infantile glaucoma. The frequency of this disease is 1 in 12,000 to 18,000 and it represents 1% to 5% of all glaucomas.16 It is characterized by isolated trabeculodysgenesis and is not associated with other ocular or systemic diseases. Seventy-five percent of cases occur bilaterally, and 65% occur in males. More than 80% of cases present before 1 year of age.
Sporadic occurrence is observed in approximately 90% of cases. The remainder demonstrate an autosomal recessive pattern with variable penetrance or a polygenic inheritance pattern.17,18 Siblings of an affected child should be examined for glaucoma. Advances have recently been made in the identification of genetic mutations associated with congenital glaucoma, including two chromosomal localizations on 2p21 (GLC3A) and 1p36 (GLC3B).19,20,21,22 For some CLC3A-linked families, various mutations in the cytochrome P4501B1 (CYP1B1) gene are found.23,24 One theory is that CYP1B1 is involved in the metabolism of a molecule important for the normal development and function of the anterior segment.19
Primary congenital glaucoma presents clinically with epiphora, photophobia, and blepharospasm. Examination often reveals increased corneal diameter, corneal cloudiness (Fig. 12), and breaks in Descemet’s membrane. Other findings include an intraocular pressure greater than 21 mm Hg, isolated trabeculodysgenesis on gonioscopy, and increased optic nerve cupping.
 Fig. 12 Primary congenital glaucoma. Buphthalmos and corneal clouding. |
A differential diagnosis of these signs and symptoms should be considered before the physician diagnoses primary congenital glaucoma. Other causes of similar corneal changes include megalocornea, metabolic diseases, corneal dystrophies, trauma, and keratitis. Epiphora or photophobia may occur in nasolacrimal duct obstruction, keratitis, iritis, trauma, and corneal dystrophies. Optic nerve anomalies simulating a glaucomatous nerve include optic pits, colobomas, and hypoplasia. Generally it is not difficult to make the diagnosis of primary congenital glaucoma when all of the signs and symptoms are considered together.
Surgery is the preferred treatment for primary congenital glaucoma. There is generally a poor response to medications and numerous potential side effects in infants. Surgery provides success rates as high as 90% and demonstrates low complication rates.
Goniotomy is the preferred surgical procedure for infants younger than 2 to 3 years of age.25,26 A single goniotomy controls intraocular pressure in the majority of cases. However, recurrence of elevated pressure occurs in 20% to 30% of cases and requires a repeat of the procedure.27 Trabeculotomy is a reasonable alternative to goniotomy for surgeons who are more comfortable with this approach and when the angle is poorly visible because of marked corneal edema. Removal of the corneal epithelium by scraping or with alcohol can improve the view for goniotomy.
LATE-DEVELOPING INFANTILE GLAUCOMA
Late-developing infantile glaucoma occurs after infancy and may be difficult to distinguish from juvenile open-angle glaucoma. It occurs in children whose intraocular pressures are not elevated enough to cause corneal clouding or enlargement but are high enough to cause optic nerve damage. The angle may have the appearance of isolated trabeculodysgenesis or show partial development of the angle recess.
GLAUCOMA ASSOCIATED WITH CONGENITAL ANOMALIES
ANIRIDIA
Aniridia is a bilateral congenital anomaly in which the iris is markedly underdeveloped (see Fig. 5). The term aniridia is a misnomer because there is generally a rudimentary iris stump of variable extent visible on examination of the angle.
Two thirds of cases are autosomal dominant with a high degree of penetrance. This form has been linked to a mutation on the short arm of chromosome 11 (PAX6 gene).28 The remainder are sporadic, and approximately 20% are associated with Wilms’ tumor.29 However, only 1% of patients with Wilms’ tumor have aniridia. A deletion of the short arm of chromosomes 11 and 13 has been demonstrated in the association of Wilms’ tumor and sporadic aniridia.30,31
Associated ocular conditions include keratopathy, cataract, ectopia lentis, foveal hypoplasia, and optic nerve hypoplasia. Optic nerve hypoplasia occurs in 75% of aniridic patients and may be related to poor macular development.32 Photophobia, nystagmus, decreased vision, and strabismus are common manifestations in aniridia. The visual acuity is generally no better than 20/200 because of the foveal hypoplasia and the accompanying nystagmus.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


