Congenital Optic Nerve Malformations
Brian P. Brook
Elias I. Trabouls
Development of the Optic Nerve
The optic nerve develops along the path of the optic stalk, which connects the optic vesicle or cup to the developing forebrain. The invagination of the optic vesicle into the optic cup leaves a ventral gap—the optic fissure—which is continuous with a groove along the ventral optic stalk. This groove transmits the hyaloid artery, which supplies blood to the developing eye. The time course over which fusion of the optic fissure occurs is quoted differently by different authors;1,2,3 most agree, however, that the closure begins at about the fifth week of gestation and is completed by approximately the seventh week of gestation. The closure of the posterior groove—which incorporates the hyaloid vasculature into the optic stalk—may not end until approximately the eighth week of gestation. Development of the optic stalk also likely depends on signals from the surrounding periocular mesenchyme.4
At about the same time as optic fissure closure, the retinal ganglion cells begin to differentiate near the edge of the nascent optic disc. Small areas of vacuolization appear in the optic stalk that allow for the transmittal of the first retinal ganglion cell axons to the diencephalon.5 The subsequent differentiation of retinal ganglion cells occurs in progressively more distal areas of the retina. This process necessitates a complex program of axonal guidance, involving both growth-promoting and growth-inhibiting signals, which vary with retinal location. Although a detailed review of these molecular signals is beyond the scope of this chapter, we discuss some of the more important molecules as they relate to specific human diseases within subsequent sections. Provis and colleagues found that, in human fetuses, the number of axons in the optic nerve reaches a maximum of 3.7 million at 16 to 17 weeks’ gestation, before undergoing a “pruning” process during which the number reduces to the adult total of 1.1 million by 29 weeks’ gestation.6,7
Neuroectodermally derived glial cells from the optic stalk envelop the hyaloid vasculature and help to delineate the early optic nerve head and the glial portion of the lamina cribrosa. Mesenchymal cells later migrate to this area and also contribute to the formation of the lamina cribrosa.
At approximately 7 months’ gestation, myelination of the optic nerve begins at the optic chiasm and progresses towards the eye. This process halts at the optic nerve head at approximately 1 month after birth, resulting in a thin layer of myelin that slowly thickens throughout childhood.
Optic Nerve Aplasia
Clinical Presentation
Aplasia of the optic nerve is a rare condition in which the optic nerve fails to develop. The precise definition of aplasia varies among authors,8,9,10 and some reported cases may actually be extreme hypoplasia of the optic nerve. However, the following diagnostic criteria generally are accepted: no pupillary light reflex and absence of the optic disc, nerve fiber layer, and retinal blood vessels on examination.11 Optic nerve aplasia is usually unilateral and may be associated with other ocular malformations, such as uveal coloboma and microphthalmia. Some patients are otherwise healthy, whereas others have central nervous system (CNS) abnormalities. Gender predilection does not appear to be present
Pathology and Etiology
In their histopathologic examination of 13 eyes with optic nerve aplasia, Weiter and colleagues noted a vestigial dural sheath entering near the normal optic nerve position; abnormal vascular anatomy with no retinal blood vessels; absence of retinal ganglion cells; and retinal dysplasia and rosette formation.9
The precise embryologic mechanism of optic nerve aplasia is unclear. Scheie and Adler originally postulated that failure of mesenchyme to enter the optic fissure would cause a premature closure of the posterior optic groove and secondary failure of optic nerve development. Yanoff asserts that this case is actually extreme optic nerve hypoplasia.10,12 Weiter et al. disputed this theory, noting that the dural sheath is present in these eyes and is of mesenchymal origin (9). Yanoff et al. propose that the initial abnormality is a failure of the ganglion cells to develop.10 The failure of mesenchymal elements such as retinal blood vessels and the atrophy of the optic stalk would, therefore, be secondary
Prognosis and Treatment
No specific therapy is indicated for optic nerve aplasia; it is a static condition. Many of these eyes may be enucleated, either because of secondary complications or because a superior cosmetic result can be obtained with an orbital implant and shell.
Optic Nerve Hypoplasia
Clinical Presentation
Optic nerve hypoplasia (ONH) is a congenital, nonprogressive developmental abnormality characterized by reduced numbers of retinal ganglion cells and corresponding defects in the nerve fiber layer of the retina.13,14,15,16 These small optic nerves frequently are surrounded by a ring of yellowish-white scleral tissue that is approximately the size of a normal optic nerve (Fig. 26.1). The edges of the actual nerve tissue and the ending of the RPE and choroidal layers form the so-called “double-ring sign,” which is not invariably present. The severity of the hypoplasia is extremely variable (Fig. 26.2), and tortuosity of the retinal blood vessels may be present. Variable amounts of peripapillary and nerve pigmentation also are observed. ONH may be unilateral or bilateral;17,18 it is usually isolated, but may be associated with other brain abnormalities, such as anencephaly or hydranencephaly. In their series of 93 patients with ONH, Skarf and Hoyt found delayed development in 23 children, most of whom had bilateral involvement with poor visual acuity and nystagmus.19
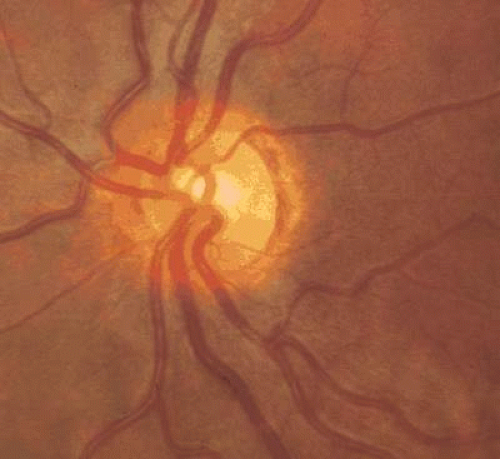 Figure 26.1. Mild optic nerve hypoplasia. Note external rim of what may be interpreted as edge of optic nerve head. A faint double-ring sign is present. |
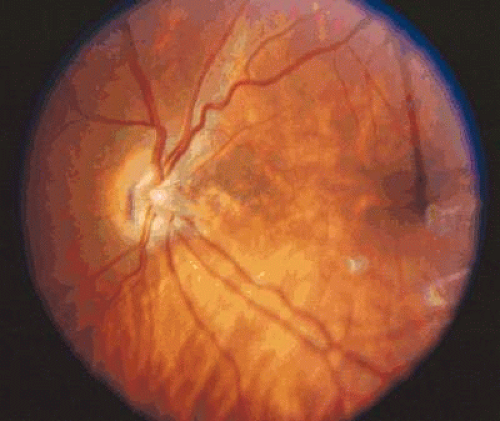 Figure 26.2. Moderate hypoplasia of the optic nerve with mild retinal vascular tortuosity. |
Patients with ONH may present with reduced visual acuity, amblyopia, strabismus, or nystagmus (in bilateral cases). Although visual acuity is usually subnormal, good visual acuity has been reported.20 The pupils may be sluggish and—in asymmetric or unilateral cases—show an afferent pupillary defect. The foveal reflex may be blunted. A variety of visual field defects, including binasal, bitemporal, arcuate, cecocentral, and altitudinal defects have been observed. Some patients, especially those with severe ONH, have foveal hypoplasia (Fig. 26.3)
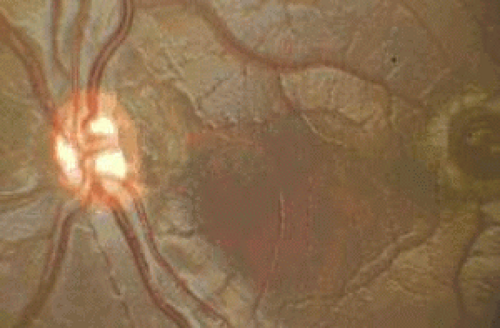 Figure 26.3. Severe optic nerve hypoplasia and foveal hypoplasia. |
Associations between multiple systemic conditions and syndromes and ONH have been reported (Table 26.1). Note that some of these associations have been noted in relatively few patients—sometimes only a single case report—whereas other associations (e.g., fetal alcohol syndrome, Goldenhar syndrome, Duane syndrome) are more established.21,22,23,23,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43 When accompanied by an absent septum pellucidum (Fig. 26.4) and thinning or agenesis of the corpus callosum and pituitary dysfunction, ONH is referred to as septo-optic dysplasia or de Morsier syndrome.44 Pituitary dysfunction is relatively common in these patients. Although growth hormone deficiency is the most common endocrine abnormality,45 central hypothyroidism, elevated prolactin, neonatal hypoglycemia/adrenocorticotropic hormone (ACTH) deficiency, diabetes insipidus, trophic hormone hypersecretion, early-onset or delayed puberty, abnormal thermoregulation and pan-hypopituitarism have been reported.19,46,47,48,49,50 A history of neonatal jaundice (indicative of hypothyroidism) and/or neonatal hypoglycemia (indicative of adrenal insufficiency) may be elicited.
Table 26.1. Systemic disorders in which optic nerve hypoplasia has been reported. | |
|---|---|
|
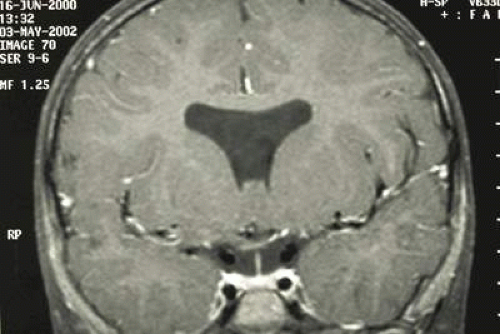 Figure 26.4. Absence of the septum pellucidum in a patient with septo-optic dysplasia. |
Magnetic resonance imaging (MRI) is the study of choice to detect associated central nervous systemic pathology. In their series of 15 patients with ONH, Brodsky et al. describe thin, signal-attenuated optic nerves and varying degrees of chiasmal hypoplasia.51 In addition to the classic abnormalities of septo-optic dysplasia, posterior pituitary ectopia, hemispheric migration abnormalities, and intrauterine/perinatal cerebral injuries may be observed in a considerable proportion of these patients.52 Pituitary ectopia and hemispheric abnormalities appear to be good indicators of developmental delay and endocrine dysfunction. An isolated septum pellucidum, in general, is not associated with significant comorbidities.
Among children in U.S. schools for the blind, between 4% and 13% have ONH.53,54 Among the participants of the Tajimi Eye Health Care Project in Japan, approximately 0.3% of those examined had superior segmental ONH (i.e., asymmetric hypoplasia affecting mainly the superior portion of the optic nerve).55 Approximately 9% of children born to mothers with type I diabetes mellitus are reported to have superior segmental ONH.56 Rudanko and Laatikainen found that, in Finland, the population prevalence of ONH was 1.5 per 100,000; it accounted for approximately 4% of visually impaired children.57 A small pilot study in Sweden suggests that the percentage of visually impaired children with ONH may be greater today than it was in the past.58
Pathology and Etiology
Histopathologic examination of eyes with ONH have revealed two different configurations of the optic nerve head.11 In those eyes with a classic double-ring sign, the retinal pigment epithelium (RPE) extends over the outer portion of the optic nerve head, with only a small, central area through which the retinal vessels entered the eye.13 The outer ring observed clinically represents the junction between the sclera and the lamina cribrosa; the inner ring represents the termination of the RPE. A second configuration, in which no double-ring sign was present, consisted of a normal termination of the RPE. The number of ganglion cells varies from absent to near normal, whereas the nerve fiber layer is reduced to absent, often populated only by glial elements. Retinal vasculature is generally normal on examination, although a reduced number of vessels have been observed and some hyaloid remnants noted. We should note that many of the reported cases for which histopathology is available are from stillbirths or children with other developmental or CNS abnormalities. Thus, the reported observations may not be representative of ONH as an isolated finding.
The pathogenesis of ONH is not understood completely. Most cases are sporadic and, as such, could have genetic, stochastic, and/or environmental causes. In general, one could postulate that ONH results from either (a) an initially reduced number of retinal precursor cells becoming retinal ganglion cells and/or (b) an abnormal destruction of these cells after they initially differentiate.
A number of prenatal exposures have been associated with ONH (Table 26.2).29,59,60,61,62,63 ONH is a relatively common feature of fetal alcohol syndrome. A now-classic association between superior segmental hypoplasia of the optic nerve (SSHON) and maternal diabetes was first reported by Peterson and Walton in 17 children with altitudinal visual field defects and relatively good visual acuity.64 Clinical features that aid in the diagnosis of SSHON include: relatively superior entrance of the central retinal artery, pallor of the superior disc, a superior peripapillary halo, and thinning of the superior peripapillary nerve fiber layer.65 The prevalence of SSHON in children of type 1 diabetic mothers was estimated by Landau et al. to be 8%.56 The sample size in this study, however, was 34 patients. Therefore, the precise prevalence could be higher or lower than this estimate.66. Not all patients with SSHON, however, are born to mothers with diabetes.67 We have observed a patient with an inferior segmental hypoplasia of the optic nerve (Fig. 26.5).
Table 26.2. Drugs reported in association with optic nerve hypoplasia | |
|---|---|
|
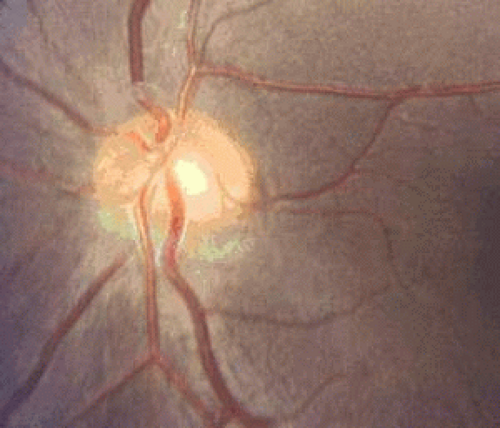 Figure 26.5. Inferior sectoral hypoplasia of the optic nerve head. |
Most recent work on the genetics of ONH has centered on molecules important in axonal guidance and on transcription factors important in ocular and CNS development. As previously mentioned, a complex network of guidance cues is required for retinal ganglion cell axons to find their way into the optic nerve head and to the lateral geniculate nucleus. One such molecule, netrin-1, is expressed in the neuroepithelial cells at the optic disc and binds to a cell surface receptor, DCC, on retinal ganglion cells.68 Although mouse embryos lacking either DCC or netrin-1 show normal growth of ganglion cell axons toward the optic nerve, these axons fail to enter the optic nerve, resulting in ONH. The fact that some axons do make it into the optic nerve suggests that other signaling cues also may be at work. Mutation of netrin-1 in mouse also disrupts the development of other midline structures such as the corpus callosum and the hypothalamus.69,70 To date, no mutations in either netrin-1 or DCC have been reported in humans with ONH.
The paired-like homeobox transcription factor gene Hesx1 is dynamically expressed in midline structures of the developing mouse embryo, including the Rathke pouch (the pituitary primordial), the ventral prosencephalon, and the diencephalon.71 Mice lacking Hesx1 show anophthalmia or microphthalmia and defects in the prosencephalon, olfactory system, corpus callosum, and other midline brain structures. Dattani et al. identified two siblings with septo-optic dysplasia and homozygous mutations in HESX1 that destroy DNA binding of the protein. Thomas et al. describe three heterozygous mutations in well-conserved codons in patients with milder variants of septo-optic dysplasia and congenital pituitary hypoplasia, suggesting that haploinsufficiency for this transcription factor also may produce a phenotype.72 Last, Tajima et al. have reported a patient with a heterozygous mutation in HESX1 associated with left ONH and mild combined pituitary hormone deficiency.73 The precise role that HESX1 plays in regulating retinal ganglion cell axon growth is a matter currently under investigation.
The paired homeobox transcription factor PAX6 has also been shown to be mutated in some patients with posterior segment abnormalities. Azuma et al. reported mutations in the transcription factor PAX6 in four patients with varying degrees of ONH.74 We should emphasize that the absolute number of patients with demonstrable mutations in HESX1 or PAX6 is small compared with the number of patients with ONH; this suggests that additional genes and additional mechanisms are likely to be discovered.
Oster and colleagues postulate that a relationship may exist between the role of insulin- or insulin-like growth factor (IGF) receptors in the pathogenesis of superior segmental ONH.5 Although this is currently a hypothesis under investigation, they cite evidence that insulin receptors are used elsewhere in the brain for axonal pathfinding and that insulin- and IGF-receptors are found in the developing retina
Prognosis and Treatment
The reduction of vision in ONH is generally nonprogressive, although amblyopia may be superimposed on the visual loss of ONH per se.75 Correction of refractive errors and a trial of patching is therefore recommended.
Perhaps the most important intervention in ONH is accurately diagnosing associated endocrinopathies or CNS abnormalities in a timely manner. A recognition of endocrine comorbidities may prompt earlier treatment and avoid complications such as sudden death and perioperative seizures.76,77 Because hyperprolactinemia may stimulate growth transiently, even in the absence of detectable growth hormone, an endocrine work-up is still warranted in young children, even in the setting of normal growth patterns.49
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


