The Corneal Dystrophies
Glenn C. Cockerham
Kenneth R. Kenyon
Corneal dystrophies typically exhibit a familial pattern, are bilateral and generally symmetric, and are not known to be caused by environmental or systemic factors. Most corneal dystrophies present relatively early in life, with exceptions of Fuchs endothelial corneal dystrophy and the iridocorneal endothelial syndrome, and are variably progressive. They may affect any layer of the cornea, or multiple tissues. Inflammation may occur late in the course but is not an initial manifestation. The central cornea is generally affected more than the periphery, with the exception of Bietti crystalline corneal dystrophy.
Insights into the molecular genetics, biochemistry, and clinical variants of corneal dystrophies have greatly increased within the last decade. These developments build upon a large existing knowledge base of clinical genetics and pathology to improve diagnosis and management. Likewise, new options, such as excimer laser surgery, are available for management of anterior dystrophies. In this chapter terminology for the various dystrophies reflects current usage within the ophthalmology literature. Eponymous terminology for the dystrophies is also included. Similar to the previous edition, the dystrophies are classified by the corneal tissue predominantly affected, from anterior to posterior (Table 1). This approach does not necessarily correspond to the possible pathogenetic mechanisms, for instance, epithelial cells producing the abnormal products found within some stromal dystrophies. However, identification based on slit lamp appearance is familiar to most ophthalmologists and provides a framework for discussion.
TABLE 1. Cornea Dystrophies | |
|---|---|
|
EPITHELIAL DYSTROPHIES
Anterior Basement Membrane Dystrophy
Anterior basement membrane dystrophy (ABMD), also known as map-dot-fingerprint or Cogan microcystic dystrophy, is the most common anterior dystrophy. It is commonly found by the ophthalmologist who seeks it.1,2,3 Inheritance is considered to be autosomal dominant, although many patients with this condition deny other family involvement.4 Guerry initially described patients with fine clusters of fingerprintlike lines in the basal epithelium and later described patients with maplike inclusions.5,6 Cogan described grayish white cysts within the corneal epithelium1 (Fig. 1). Signs of ABMD may be inapparent on slit lamp biomicroscopy but become more visible on retroillumination. The irregular surface associated with ABMD is readily seen after application of fluorescein to the corneal surface; irregularities appear as dark-staining (“negative staining”) elevations. The majority of patients with ABMD are asymptomatic and unaware of the condition. Epithelial erosions associated with this condition have been well-described.3,7 Recurrent erosion, usually presenting as uniocular pain and foreign body sensation upon awakening, was reported in 34% of patients in one study.8
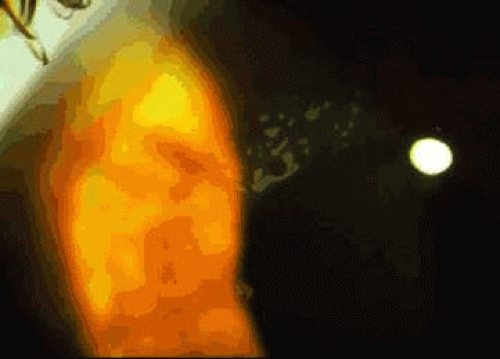 Fig. 1. Anterior basement membrane dystrophy. Gray puttylike inclusions are present within the central epithelium. These inclusions are due to duplicated epithelial basement membrane. |
Confocal microscopy reveals increased deposition of abnormal extracellular matrix in the anterior stroma.9 Histologic and ultrastructural studies have demonstrated anomalies at the epithelium-Bowman layer interface, including accumulation of basement membrane material and collagen fibers.10 ABMD and recurrent erosion syndrome appear to have similar pathophysiology.11 Incompetence of the basement membrane adhesion complexes is central to initiation and maintenance of erosions.12
Mild erosions are treated with 2% or 5% sodium chloride drops during the day and 5% sodium chloride ointment or other mild ointment at night. This regimen is usually continued for at least 1 to 2 months. Dry eye and meibomian gland inflammation contribute to recurrent erosions and should be treated with ocular lubrication and warm compresses. Topical steroids in conjunction with oral tetracycline reduce recurrent erosion, presumably through decrease in metalloproteinase-9.13,14 Bandage soft contact lenses are useful for pain control in acute erosions, and continuous wear of soft contact lenses for 4 to 6 weeks may allow adequate healing of the ocular surface, with restoration of epithelial adherence. Surgical options to facilitate epithelial adherence include anterior stromal puncture (ASP), superficial keratectomy, superficial keratectomy with diamond burr polishing of the Bowman layer, and phototherapeutic keratectomy (PTK) or photorefractive keratectomy (PRK). ASP consists of punctures through epithelium into anterior stroma under topical anesthesia with a bent needle tip or other sharp instrument, avoiding the visual axis. The resultant small scars help seal the treated areas.15,16 Pathologic studies of ASP demonstrate activated keratocytes, new basement membrane with type IV collagen, and production of new fibronectin and laminin.17,18,19 Superficial keratectomy is useful to remove loose epithelium with a cotton swab or surgical blade; loose epithelium is easily removed up to an area of firmer attachment. Surgical stimulation appears to permit better adherence after final healing. Polishing of the Bowman layer with a diamond burr may reduce the rate of recurrent erosion and improve vision.20,21 A bandage contact lens and topical antibiotics are required until full epithelial coverage of the surgical defect is achieved. Lastly, removal of central epithelium and/or Bowman layer with an excimer laser by PTK or PRK appears to be safe and effective and may lead to better epithelial adherence. Recurrent erosions after PTK may require additional treatments.22,23,24,25 Patients interested in refractive surgery must be carefully evaluated for the presence of ABMD. Due to inadequate epithelial adhesion, patients with ABMD are at risk for poor flap formation due to slippage or “skipping” of the microkeratome head during laser-assisted in situ keratomileusis (LASIK). Surface ablation techniques are preferred in this setting.26
Meesmann Epithelial Corneal Dystrophy
Meesmann epithelial corneal dystrophy (MECD), also known as juvenile hereditary epithelial dystrophy, is a relatively mild bilateral dystrophy affecting cornea-specific keratins. Keratins are intermediate filaments found in various epithelia that are expressed in pairs, consisting of a type 1 (keratins 9–21) and type 2 (keratins 1–8) protein. Corneal epithelial cells specifically express keratins 3 (K3) and 12 (K12).27 Mutations within the genes coding for these keratins in patients with MECD were initially described by Irvine et al. in 1997.28 Further mutations within the K12 gene, located within chromosome 17q12, as well as the K3 gene have been reported in MECD.29,30,31,32,33 Inheritance is autosomal dominant.
Patients with clear intraepithelial microcysts characteristic of MECD were initially described by Pameijer; a larger series was reported later by Meesmann and Wilke.34,35 Stocker and Holt published the first cases in the English literature.36 Most families described are of European origin; the dystrophy has also been reported in patients of Saudi Arabian ancestry.37 Cysts typically appear in infancy, within the interpalpebral areas, and increase in number with age. Bilaterally symmetric cysts are the norm, although asymmetric or possibly unilateral involvement is possible.38 The microcysts appear gray on direct illumination and transparent by retroillumination (Fig. 2). Punctate staining of the cornea by fluorescein reveals ruptured cysts. By confocal microscopy, the cysts appear as hyporeflective areas measuring 48 to 145 μm in diameter.39 Corneal sensation typically remains intact. Painful erosions may occur later in life, with subepithelial scarring.40,41,42
 Fig. 2. Meesman epithelial corneal dystrophy. Clear microcysts within the epithelium are visible. These cysts contain “peculiar substance” due to mutations within the gene coding for epithelial keratins. |
Histopathologic examination demonstrates disorganized epithelium containing periodic acid-Schiff (PAS)–positive cysts. Electron dense fibrogranular “peculiar substance” is a characteristic finding by transmission electron microscopy. The epithelial basement membrane may be thickened or discontinuous.43,44,45,46 Intracytoplasmic inclusions may contain keratin. Superficial keratectomy, lamellar keratectomy, or penetrating keratoplasty may be employed for corneal scarring or recurrent erosions; recurrence is common.47
Lisch Corneal Dystrophy
Lisch corneal dystrophy (LCD) is characterized by band-shaped and whorled microcysts within the corneal epithelium.48,49,50,51 The inheritance pattern described is compatible with either X-chromosomal or pseudo–autosomal dominant transmission. Linkage with chromosome Xp22 has been identified.52 On clinical examination, opaque gray lesions are present within the corneal epithelium. These lesions resemble whorls, bands, or strands, separated by clear areas. Densely packed clumps of microcysts within the opacities are evident on retroillumination. Vision is affected if the lesions occur within the visual axis. Corneal erosions are infrequent. Light and electron microscopy demonstrate cytoplasmic vacuolization of all epithelial cells within the affected areas. Visual may improve after superficial keratectomy to remove affected epithelium.
CORNEAL DYSTROPHIES OF THE BOWMAN LAYER
Corneal dystrophies which primarily affect the Bowman layer (CDB) have recently undergone reclassification. Previous reports and pathologic material were reviewed by Küchle et al., who proposed that the dystrophy reported by Reis in 1917, and subsequently described by Bücklers in 1949, be renamed corneal dystrophy of Bowman layer type 1 (CDB 1).53,54,55 The anterior corneal dystrophy described by Thiel and Behnke in 1967 has been classified as CBD II.56 Based on clinical, pathologic, and ultrastructural characteristics, many cases previously classified as Reis-Bücklers dystrophy were placed in the CDB II category. The anterior corneal dystrophy described by Grayson and Wilbrandt does not fit into either classification, although it does resemble CDB II clinically.57 Both types of CDB are inherited in an autosomal dominant manner. Patients with both types suffer from recurrent erosions and progressive visual loss. Abnormality of the corneal epithelium appears central to the pathogenesis of dystrophies of the Bowman layer.58,59
Corneal Dystrophy of Bowman Layer Type 1
Initially reported by Reis in 1917, Bücklers in 1949 described families with a hereditary corneal dystrophy of epithelium and Bowman layer and recurrent erosions.53,54 The erosions are bilateral and extremely painful and may begin in infancy, but almost always by early childhood.60 Minor trauma may precipitate erosions, or they may occur during sleep. Examination reveals geographic gray white opacification affecting the Bowman layer within the central cornea. Progressive opacification occurs with increasing visual loss, which is reported greater in CBD 1 than CBD 2.55,60,61 A mutation at R124L within the transforming growth factor beta–induced (βig-H3) gene on chromosome 5q31 has been reported.59 Other autosomal dominant corneal dystrophies with varying phenotypes, including granular dystrophy, lattice dystrophy, and mixed granular-lattice dystrophy (“Avellino dystrophy”) are also associated with mutations within this gene.62,63,64,65
Absence of the Bowman layer is noted by light microscopy, with irregularity of epithelial thickness and deposition of fibrous material within the basal layer of epithelium and the anterior stroma. The deposits stain brightly with Masson trichrome stain, similar to the deposits of granular corneal dystrophy. By transmission electron microscopy (TEM) rodlike bodies resembling those found in granular dystrophy are seen within the deposits.55,59 Hemidesmosomes and basal epithelial attachments are absent in areas. The posterior stroma and endothelium are unaffected.66,67,68,69,70,71 CDB 1 may represent a superficial variant of granular corneal dystrophy. Lamellar keratectomy, phototherapeutic keratectomy, and penetrating keratoplasty may be indicated for painful recurrent erosions or visual loss to surface irregularity and opacity.68,72,73 Recurrence after surgical treatment is common in CDB 1 and may occur as early as one year.74,75,76
Corneal Dystrophy of Bowman Layer Type II
Thiel and Behnke described patients with “honeycomb” opacities within the central cornea at the level of the Bowman layer.77 Although originally considered a subtype of Reis-Bücklers corneal dystrophy, this dystrophy is now considered distinct and classified as CDB II.55,59 Similar to CDB I, painful corneal erosions and progressive visual loss are common, although vision tends not to be as severely impaired as in CDB 1. A gene mutation at R555Q locus within the βIG-H3 gene on chromosome 5q31 has been described,59 as well as linkage of some cases to chromosome 10q23–q24.78 Pathologically, partial absence of epithelial basement membrane is seen. There is absence of the Bowman layer, with “sawtooth” fibrous tissue interposed between epithelium and stroma.55 Masson trichrome stains this fibrous tissue inconsistently, unlike the strong staining present in CDB I. Areas of lipid deposits are noted within the stroma.79 TEM demonstrates absence of hemidesmosomes and epithelial basement membrane. Subepithelial accumulations of unusual “curly” fibers are present.55,59,70 Treatment options for erosions and visually significant opacity are the same as for CDB I. Recurrence is less common within grafts, compared with CDB I.
STROMAL DYSTROPHIES
Granular Corneal Dystrophy
Granular corneal dystrophy (GCD) (Groenouw type 1 dystrophy) is considered to be the most common stromal dystrophy, usually affecting the central subepithelial area and anterior stroma. Early investigators noted “hyaline” deposits in both granular and lattice corneal dystrophies, speculating that these were variants of the same condition. It was not until 1961 that Seitelberger identified amyloid in lattice corneal dystrophy, distinguishing the two major stromal dystrophies by the biochemical features of their deposits.80 Granular corneal dystrophy is inherited in an autosomal dominant manner. It shares a genetic locus, the transforming growth factor beta–induced (βig-H3) gene on chromosome 5q31, with other autosomal dominant corneal dystrophies, including CBD type 1 (Reis-Bücklers dystrophy), lattice corneal dystrophy, and mixed granular-lattice (“Avellino”) dystrophy.63 Numerous missense mutations have been described within the βIG-H3 gene.64,65,81,82,83,84,85,86,87,88,89,90,91,92,93,94
Central involvement of the anterior corneal stroma with discrete opacifications is typical of GCD. The areas between the deposits are generally clear, as is the corneal periphery (Fig. 3). Histopathologic examination of classic GCD demonstrates discrete subepithelial and stromal deposits, which are eosinophilic with hematoxylin-eosin, stain red with Masson trichrome and are negative with PAS, Alcian blue, and colloidal iron stains.95 (Fig. 4). Keratoepithelin deposits are demonstrated in corneas with GCD by immunohistochemistry.96 TEM reveals electron-dense crystalline rods within the deposits.97,98
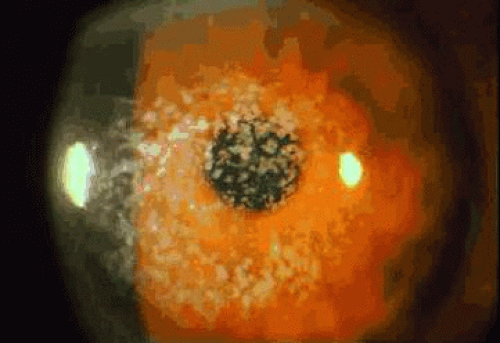 Fig. 3. Granular corneal dystrophy. This autosomal dominant dystrophy is characterized by discrete stromal opacities with intervening clear areas. Mutations within the βIG-H3 gene are associated with this condition, as well as with lattice dystrophy and corneal dystrophy of the Bowman layer. |
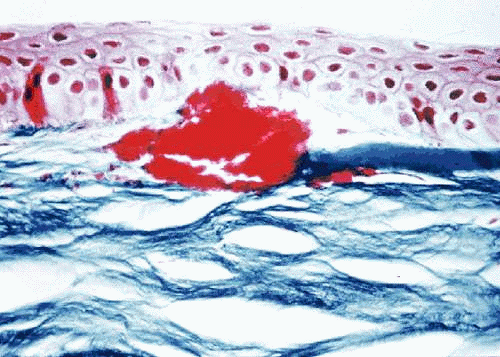 Fig. 4. Granular corneal dystrophy. Light microscopy demonstrates discrete stromal deposits within the anterior corneal stroma. Destruction of the Bowman layer is present. Masson trichrome stain. |
Treatment options are surgical. A lamellar graft may be useful for removal of involved anterior stroma if the deeper cornea is relatively clear, avoiding the risk of endothelial graft rejection. PTK is also capable of removing granular deposits within the anterior stroma, although a hyperopic shift is induced by the procedure.99 Penetrating keratoplasty is indicated for visually significant deposits not amenable to lamellar keratoplasty or PTK. Recurrence within grafts is common, commencing from the graft-host interface.100 Recurrent deposits may be superficial or deep.100,101,102,103
Lattice Corneal Dystrophy
Lattice corneal dystrophy (LCD) (Biber-Haab-Dimmer dystrophy) is characterized by deposits of amyloid proteins within the corneal stroma. LCD was initially described by Biber in1890, with later reports by Haab and Dimmer.104,105,106 As mentioned above, early investigators commented on hyaline inclusions within the corneal tissue, noting that granular and lattice corneal dystrophies shared some similarities. Seitelberger discovered the characteristic amyloid inclusions in LCD.80
Classic lattice dystrophy (type 1 LCD) usually presents by age 20 with visual decrease and recurrent erosions. Examination of affected children may detect changes of LCD by age three. Early changes include subepithelial round or ovoid white opacities and axial anterior stromal haze. Delicate filaments, which appear refractile on retroillumination and white on direct illumination, are noted within the corneal stroma (Fig. 5). The disease is localized to the cornea, with no associated abnormalities.107,108,109 In LCD type II, first described by Meretoja in 1969 in a large series of Finnish patients, later onset of dystrophic changes between 20 and 35 years is common, with fewer symptomatic erosions. Lattice lines tend to be fewer and thicker and appear to extend from the periphery toward the center. Good vision is often maintained until late in life. Transmission is autosomal dominant. Unlike LCD type 1, systemic findings are present and include paralysis of cranial nerves, notably the facial nerve and peripheral nerves, dry skin, a “masklike” facies, autonomic dysfunction, and proteinuria.110,111,112 Although LCD type II appears to be most common in Finnish families, it has also been described in Japanese patients.113 Type III LCD is characterized by a thicker lattice deposits within the corneal epithelium, later onset (seventh to ninth decade), lack of corneal erosions, and no associated systemic anomalies.89,109 Families with LCD types IIIA, IV, VI, and VII have been described.114 LCD, like most corneal dystrophies, is usually bilateral, although unilateral or asymmetric cases have been reported.115,116 Confocal microscopy may be helpful in distinguishing amyloid deposits and nerve degeneration in LCD from other entities, such as infectious crystalline dystrophy, acanthamoeba, and fungal hyphae.117,118
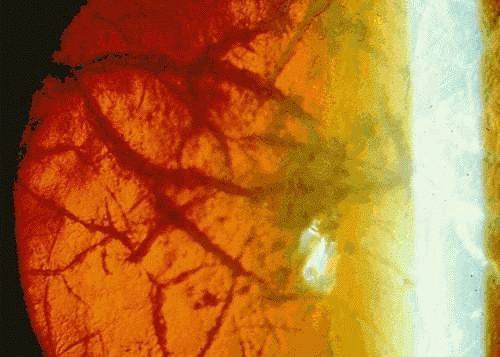 Fig. 5. Lattice corneal dystrophy. This autosomal dominant dystrophy, also associated with mutations of the βIG-H3 gene, is evident as branching “latticelike” opacities within the stroma, as well as subepithelial deposits, best appreciated on retroillumination. |
LCD type 1 is associated with mutations in the transforming growth factor beta–induced (βig-H3) gene on chromosome 5q31, whose normal product is keratoepithelin. Other autosomal dominant corneal dystrophies with varying phenotypes, including granular dystrophy, mixed granular-lattice dystrophy (“Avellino dystrophy”) and Reis-Bücklers dystrophy are also associated with mutations within this gene (see section on granular dystrophy above). Type II LCD (Meretoja) is due to mutations within the gelsolin gene on chromosome 9q34.119,120,121,122 LCD types IIIA, IV, VI, and VII are all related to point mutations within the βig-H3 gene.114
Histopathologic examination demonstrates linear and nodular eosinophilic deposits located within the stroma and underneath the epithelium. These are poorly visualized with routine hematoxylin-eosin staining; however, the lesions are congophilic and revealed as distinct amyloid deposits with Congo red staining (Fig. 6). Another feature of amyloid protein is birefringence of the deposits under polarized light after staining with Congo red. Amyloid also stains with PAS, is metachromatic with crystal violet, and fluoresces with thioflavin T staining.107,123,124,125,126
 Fig. 6. Lattice corneal dystrophy. Histopathologic examination reveals the presence of amyloid within the corneal stroma, detectable by several methods. A: Amorphous pink deposits are visible by periodic acid-Schiff stain (PAS). B: The amorphous areas stain red with Congo red and demonstrate “apple green” birefringence with polarized light. |
The composition of the amyloid protein differs within the major types of LCD. Immunohistochemical studies have revealed protein AA and protein AP within amyloid deposits of LCD type I.127,128 Amyloid deposits in type II LCD (Meretoja) have been reported to stain with antibodies against transthyretin-related amyloid fibril whole protein isolated from myocardium of a patient with familial amyloid polyneuropathy, as well with protein AP, but not protein AA.129 In another study of type II LCD immunostains against proteins AA and AP were negative.130 Amyloid deposits in LCD type III stain demonstrate strong immunostaining for protein AP and weak or absent immunostaining for protein AA.129 Immunohistochemical studies reveal that the amyloid deposits of families with LCD types I and IIIA contain fragments of keratoepithelin.96,131
Since deposits occur centrally, vision loss is common. Penetrating keratoplasty remains the treatment of choice. Recurrences may occur in up to 48% of patients.132,133 Recurrences may primarily affect the anterior stroma occurring as early as 3 years or later than 20 years after the initial graft.133,134 PTK may be helpful in cases of primary or recurrent LCD affecting the anterior stroma.135
Mixed Granular-Lattice Dystrophy (Avellino)
Several studies reported the presence of amyloid within the corneas of patients with granular corneal dystrophy.97,136 Folberg described the presence of amyloid within the corneas of patients with clinical and histologic granular dystrophy.137 The clinical presentation was atypical for classic GCD, with stromal haze between the granular deposits. Each affected patient traced ancestry to the Italian province of Avellino. Later studies confirmed these findings.138 Similar to granular and lattice corneal dystrophies, transmission is autosomal dominant and localizes to the transforming growth factor beta–induced (βig-H3) gene on chromosome 5q31.139 Like GCD and LCD, keratoepithelin deposits are found in Avellino dystrophy by immunohistochemistry.96 Penetrating keratoplasty is performed for visually significant involvement. Recurrence within the graft is possible. LASIK is contraindicated in Avellino dystrophy, due to increased deposits within the interface and posterior stroma after surgery.140,141
Macular Corneal Dystrophy
Macular corneal dystrophy (MCD) (Groenouw type 2 dystrophy) is characterized by deposits of acid mucopolysaccharides within the corneal epithelium, stroma, and endothelium. Jones and Zimmerman recognized the nature of these deposits within the stroma by histochemical techniques.95,142 Klintworth and Vogel emphasized this dystrophy as an intracellular storage disease due to a metabolic defect.143 Experimental studies demonstrate impaired synthesis of keratin sulfate, a glycosaminoglycan normally present in the stromal extracellular matrix, with increased production of chondroitin-6-sulfate and possibly other glycosaminoglycans by keratocytes.144,145,146,147 In MCD type 1, neither the cornea nor the serum contains measurable amounts of sulfated keratin, whereas both contain sulfated keratin in type II MCD.148
Transmission is autosomal recessive; as such, MCD is uncommon in the general population, but more common in areas with isolated gene pools, such as Iceland, or areas with consanguinity. MCD is linked to chromosome 16 (16q22).149,150,151 Recent studies have identified a variety of mutations in a carbohydrate sulfotransferase gene (CHST6) encoding corneal glucosamine N-acetyl-6-sulfotransferase.152,153,154,155,156
Diffuse superficial clouding becomes evident within the central, superficial stroma in the first decade of life, eventually involving the entire cornea to the periphery (Fig. 7). Central corneal thinning is present in this dystrophy.157 Routine histologic examination may reveal an irregular epithelium with disruption of the Bowman layer, a fine granular appearance both within and surrounding keratocytes, and thickening of the Descemet membrane with occasional guttate excresences. Histochemical stains which identify acid mucopolysaccharides include alcian blue, colloidal iron, and PAS.136,142,143 Deposits are present throughout the cornea, involving epithelium, keratocytes, extracellular matrix, and endothelium. (Fig. 8).158 In corneal buttons removed after recurrence within a transplant, typical AMP deposits are noted; these are denser at the graft periphery than at the center.143
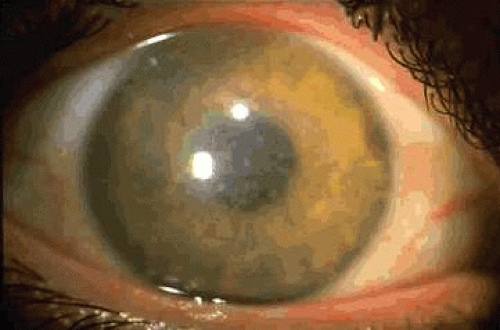 Fig. 7. Macular corneal dystrophy. This autosomal recessive dystrophy displays full-thickness stromal haze combined with focal stromal opacities. Impaired production of keratin sulfate leads to increased production of other glycosaminoglycans. |
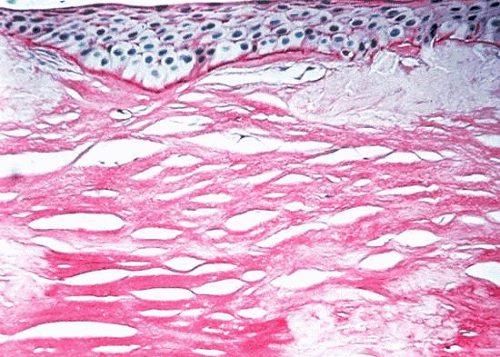 Fig. 8. Macular corneal dystrophy. Light microscopy demonstrates acid mucopolysaccharides within all layers of the cornea. Alcian blue stain. |
Corneal transplantation remains preferred therapy for MCD. In Saudi Arabia, MCD accounts for 87% of penetrating keratoplasty performed for classic corneal dystrophies.159 Recurrence accounted for 40% of the failed grafts in one study, with a mean interval of 7 years until visually significant relapse.160 However, in a different population, recurrence of MCD was less common.161
Polymorphic Stromal Dystrophy
Polymorphic stromal dystrophy represents a manifestation of corneal amyloid clinically distinct from the lattice dystrophies and gelatinous droplike dystrophy. Thomsitt and Bron described patients with a variety of posterior stromal opacities consistent with the type of dystrophic changes reported in 1939 by Pillat.162,163 Axial polymorphic opacities resembling stars and snowflakes were noted, and branching filamentous opacities were noted in the posterior cornea. The lesions were gray white and mildly refractile when viewed with direct light but were transparent in retroillumination. The intervening stroma was clear. Vision is minimally affected. Histochemistry and electron microscopy demonstrate typical amyloid deposits within the opacities.164,165 The late appearance of the opacities, the lack of progression, and the lack of familial association help to distinguish this condition from lattice dystrophy.
Gelatinous Droplike Dystrophy
Gelatinous droplike corneal dystrophy (GDLD), also known as familial subepithelial corneal amyloidosis, is characterized by deposition of large amounts of amyloid beneath the corneal epithelium. It was first reported by Nakaizumi in Japanese patients.166 GDLD remains a common dystrophy in Japan, where the incidence is estimated to be 1 in 33,000.167,168 The condition has also been described in patients of other ethnicity.169,170,171,172,173,174,175 GDLD accounted for 4.4% of corneal transplants at a center in southern India.176 Inheritance is autosomal recessive. The responsible gene has been identified as membrane component, chromosome 1, surface marker 1 (M1S1) on the short arm of chromosome 1.177,178,179,180 Several mutations within M1S1 have been reported.172,181,182
Photophobia and epiphora start within the first decade of life. Progressive vision deterioration occurs within the teen-age years. The clinical presentation initially described is a nodular deposition of yellow or white amyloid within the central cornea, eventually forming a “mulberry” configuration.169,183,184 Other presentations include band keratopathy and either localized or more diffuse opacity.180,185 Both corneas are usually affected, although a predominantly unilateral case has been reported.186 Corneal sensation may be decreased within involved areas. Stromal vascularization and scarring lead to severe vision loss. Most patients require keratoplasty by age 30.187
Histopathologic examination demonstrates an irregular epithelium, with degenerated or absent Bowman layer. Fibrils consistent with amyloid are present within basal epithelial cells by electron microscopy.188 Sheets of amyloid (PAS and Congo red positive, birefringent on polarization) are present within the anterior stroma.168,169,170,184,189 Chronic inflammatory cells and neovascularization are present within adjacent stroma in some cases. Macrophages may contain amyloid fibrils.190 Collagenous stromal scars occur in more advanced cases.191,192 Immunostains have demonstrated lactoferrin, apolipoproteins J and E, and gelsolin within amyloid deposits of GDLD.168,193,194 Abnormal corneal epithelium is implicated in the deposition of amyloid. Epithelial cells may produce amyloid fibrils, as noted above. Alternatively, increased epithelial permeability may allow tear proteins such as lactoferrin access to the anterior stroma.192,193,195
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


